PRESENTACIÓN DE CASO
Hemocromatosis hereditaria tipo I: a propósito de cuatro casos confirmados
Hereditary hemochromatosis type I: apropos of four confirmed cases
Dra. Norma D Fernández-DelgadoI; MSc Mariela ForrellatBarriosI; Dr. Roberto Valledor-TristáII; Dra. Kalia Lavaut-SánchezI; Lic. Ismael Cervera GarcíaIII
I Instituto
de Hematología e Inmunología, La Habana, Cuba.
II
Hospital General Docente "Enrique Cabrera", La Habana, Cuba
III
Centro Nacional de Genética Médica, La Habana, Cuba.
RESUMEN
La hemocromatosis hereditaria es un trastorno genético. En los últimos años se ha profundizado en el conocimiento de su fisiopatología y diagnóstico. Estos síndromes se caracterizan por sobrecarga de hierro y se distinguen varios subtipos de acuerdo con la mutación existente. Dentro de ellas, las mutaciones en el gen HFE o hemocromatosis hereditaria tipo I es la más común. Esta enfermedad tiene una gran morbilidad y mortalidad asociada a la sobrecarga del mineral. Se presentan 4 pacientes en los que por primera vez en Cuba se identificaron las mutaciones del gen HFE.
Palabras clave: hemocromatosis hereditaria, HFE, mutación C282Y, sobrecarga de hierro.
ABSTRACT
Hereditary hemochromatosis is a genetic disorder. Detailed studies on its physiopathology and diagnosis have been carried out over the last years. The syndromes are characterized by iron overload and several subtypes are distinguished according to the existing mutation. Among them, the mutations in HFE gene or hereditary hemochromatosis type I is the most common. This disease has a great morbidity and mortality associated to mineral overload. For the first time in Cuba, we report four patients with confirmed mutations in HFE genes.
Keywords: hereditary hemochromatosis, HFE, C282Y mutation, iron overload.
INTRODUCCIÓN
La hemocromatosis hereditaria (HH) es una enfermedad que se incluye en un grupo heterogéneo de condiciones clínicas asociadas a la sobrecarga de hierro. Se han descrito 5 variantes caracterizadas por mutaciones en los genes que codifican para diferentes proteínas involucradas en el metabolismo del mineral, que en su mayoría provocan alteración en la producción de la hepcidina.1,2
Las variantes de HH se denominan de acuerdo con la mutación existente. Así, las mutaciones del gen HFE se han designado como HH tipo I; las de la hemojuvelina y la hepcidina se conocen como HH tipo II o hemocromatosis juvenil A y B, respectivamente; la tipo III se relaciona con la mutación del receptor de la transferrina 2; y la IV se subdivide en la enfermedad por ferroportina o HH tipo IV clásica (sin resistencia a la hepcidina) y la verdadera sobrecarga de hierro donde hay resistencia a la hepcidina.3-7
La HH tipo I, de transmisión autosómica recesiva con penetrancia variable, es la más frecuente y se han descrito más de 30 mutaciones del gen HFE. Dentro de ellas, la sustitución de cisteína por tirosina en la posición 282 de la proteína (C282Y) es la más común y se presenta en el 80 % de los homocigóticos. Le siguen en frecuencia, la H63D (cambio de histidina por ácido aspártico) y la combinación de ambas. Otras mutaciones son extremadamente raras y algunas se han descrito en varios miembros de una familia.1,3,6,8
Las manifestaciones clínicas de la sobrecarga de hierro incluyen astenia o fatiga, artralgias, hiperpigmentación cutánea, hepatomegalia y menos frecuentemente, sobre todo como síntomas iniciales, alteraciones cardiacas (arritmias, fallo cardiaco, etc.) y trastornos endocrino metabólicos (hiperglicemia, amenorrea, trastornos de la libido, impotencia, infertilidad, hipofunción de la tiroides). 3,5,7
Los estudios de laboratorio son cruciales para el diagnóstico y en muchas ocasiones la causa de consulta. Entre las alteraciones más frecuentes están el incremento de las transaminasas y del hierro sérico (HS). La existencia de un índice de saturación de la transferrina (IS) mayor que 45 % y ferritina sérica (FS) elevada (mayor que 200 ng/mL en el sexo femenino y 300 en el masculino), son otros indicadores del exceso del mineral, que deben ser estudiados. Los estudios moleculares son los que confirman el subtipo de HH.3,9,10
En el presente trabajo se describen las características clínicas y de laboratorio de cuatro pacientes en los que el diagnóstico de HH tipo I se confirmó por primera vez en Cuba, mediante el estudio de las mutaciones HFE.
PRESENTACIÓN DE CASOS
Todos los pacientes fueron del sexo masculino, de piel blanca y el estudio molecular se realizó, a solicitud del Instituto de Hematología e Inmunología (IHI) entre los años 2009 y 2011, en el Centro Nacional de Genética Médica por el método de reacción en cadena de la polimerasa seguido por digestión enzimática.
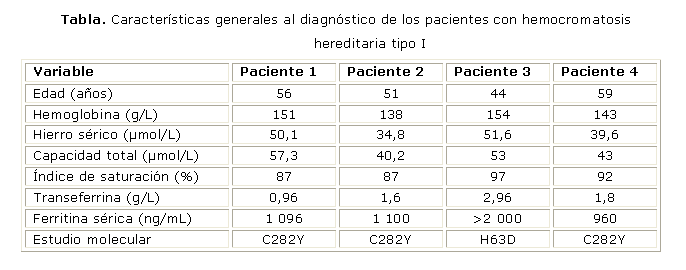
Paciente 1: 56 años, con historia de dolores articulares generalizados, incluso al reposo, edema de las manos y decaimiento. Desde 2 años antes se estudiaba por Reumatología y en un chequeo realizado se encontraron cifras elevadas de transaminasas y hierro sérico. En el hemograma se apreció, además, trombocitopenia ligera (plaquetas entre 120 y 140 x 109/L). En el IHI se le realizó estudio de sideremia (HS, capacidad total de saturación de la transferrina (CT), IS, transferrina (TF) y FS) y medulograma, Todos los exámenes confirmaron el incremento de hierro, tanto circulante como en los depósitos, por lo que se indicó el estudio molecular.
Paciente 2: 51 años, con historia de dolores articulares, diabetes mellitus tipo II, valores elevados de hierro y referencia de que le habían realizado múltiples flebotomías sin lograr la reducción del HS. Acude al IHI para realizarse estudio ferrocinético y se decidió realizar el estudio de sideremia y de las enzimas hepáticas y al comprobar los valores elevados, se indicó el estudio molecular. El estudio ferrocinético mostró un incremento moderado de la actividad eritropoyética funcional con hierro muy aumentado. Actualmente tiene diagnóstico de cirrosis realizado por biopsia hepática.
Paciente 3: 44 años, con antecedentes de tener un hermano con hemocromatosis clínica de 2 años de evolución. En dos estudios consecutivos de hemoquímica se apreció incremento de las aminotransferasas y del HS. Solo refirió descamación de la piel de manos y pies. Se realizaron los estudios de sideremia y al confirmar los valores elevados del mineral se indicó el estudio molecular.
Paciente 4: 59 años, antecedentes de diagnóstico sugestivo de hemocromatosis por los estudios de sideremia realizados en el IHI desde hacía más de 10 años, y que llevaba tratamiento con flebotomías. Asintomático en el momento del estudio molecular, pero al diagnostico tenia artralgias y hepatomegalia de 2 cm.
Los resultados de los estudios de sideremia al diagnóstico y el estudio molecular de todos los pacientes se muestran en la tabla. En los primeros tres casos todos los estudios se realizaron en tiempo cercano al estudio molecular. En el caso 4, la sideremia que se muestra correspondió al momento del diagnóstico sugestivo de HH; posteriormente, el estudio molecular confirmó el tipo de HH
En 3 pacientes se encontró la mutación C282Y y el otro fue positivo para la H63D. Todos fueron homocigóticos para la mutación detectada. En todos estuvo ausente la anemia, hepatitis aguda o daño hepático por otra causa, y ninguno refirió antecedentes de transfusiones anteriores, alcoholismo ni enfermedades oncológicas. En un solo paciente (No. 2) existió hiperglucemia ligera (glicemia en 10,2 µmol/L), lo que coincidió con sus antecedentes personales. En los cuatro se encontraron cifras de amilasa sérica entre el 5 y el 8 % sobre el valor de referencia. En otros estudios realizados (ecografía abdominal, ecocardiograma) se encontró aumento de la ecogenicidad del hígado en todos los casos, con un patrón granular grueso, bordes ligeramente irregulares y prominencia del lóbulo caudado en 2 de ellos (pacientes 1 y 2). Ninguno mostró evidencias de daño cardiaco.
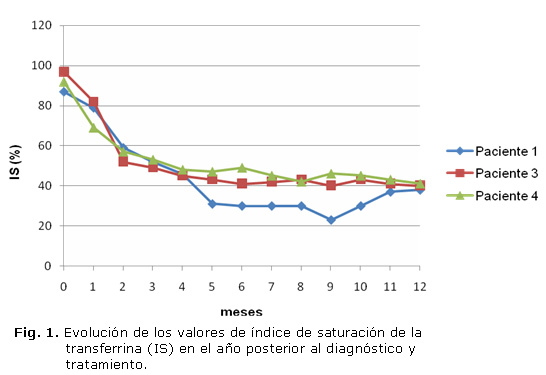
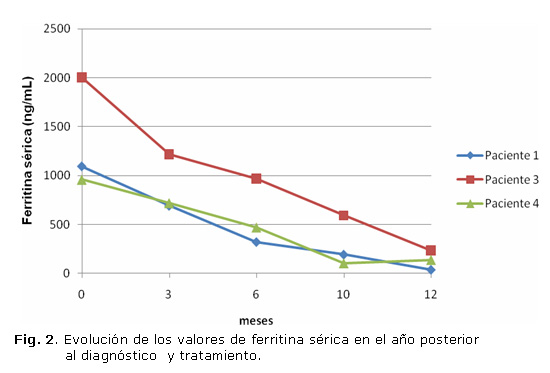
Todos fueron tratados con flebotomías semanales hasta lograr la disminución de las cifras de ferritina por debajo de 300 ng/mL, en que pasaron a realizarse quincenal, mensual y luego trimestralmente, de acuerdo con la evolución. Un paciente estuvo ausente de consulta por más de un año, luego de tener valores de FS inferiores a 50 ng/mL, y al regreso fue necesario reiniciar el régimen de flebotomías semanales (FS 496 ng/mL), valor ligeramente superior al rango de referencia de los estuches comerciales que se emplean en el centro (13 - 400 ng/mL). Las figuras muestran la evolución de los valores de IS mensuales (figura 1) y de FS trimestrales (figura 2) en los casos seguidos en el IHI (pacientes 1, 3 y 4) en el primer año posterior al diagnóstico y con tratamiento de flebotomías.
DISCUSIÓN
En los últimos 15 años, grandes cambios han ocurrido en el metabolismo del hierro lo que ha mejorado su comprensión. Se han descubierto nuevas proteínas y genes involucrados que explican los mecanismos moleculares que intervienen en la absorción del mineral, los mecanismos homeostáticos y la regulación del transporte hepático. El descubrimiento del gen HFE, localizado en el cromosoma 6p21.3-22.1, ha sido crucial para identificar las diferentes formas de HH y otras variantes genéticas de sobrecarga de hierro.2,11-13
El hierro, tan necesario para múltiples procesos en el organismo humano, resulta muy tóxico cuando está en exceso y en las enfermedades genéticas esta acumulación suele ser progresiva y provoca un grado variable de disfunción de órganos, fundamentalmente hígado, corazón, articulaciones y piel, después de varias décadas, por lo que la edad al diagnóstico suele estar entre la cuarta y quinta décadas de la vida.2,12-14
A pesar de que la población cubana se caracteriza por una gran heterogeneidad racial y sería necesario hacer estudios poblacionales para describir cómo se comportan estas mutaciones en el país, los casos estudiados hasta ahora coinciden con lo descrito internacionalmente en cuanto a tipo de mutación, predominancia de género y apariencia racial.1,3-6
El incremento de las aminotransferasas y una hepatomegalia ligera pueden ser las únicas manifestaciones en un gran porcentaje de los casos. El incremento de hierro en hígado puede estar presente entre el 38 y el 97 % de los pacientes, pero entre el 10 - 25 % desarrolla fibrosis y entre el 4 - 6 % llega a la cirrosis.10
En el 75 % de los pacientes estudiados se presentó alguna manifestación articular. La artropatía, generalmente simétrica y poliarticular se describe hasta en el 50 % de los enfermos, con afectación frecuente de las articulaciones metacarpo-falángicas.
La hiperpigmentación cutánea es una manifestación temprana de la enfermedad y se ha descrito en el 90 % de los afectados. Las uñas planas y la pérdida de los vellos del cuerpo también son frecuentes.1,10 En los pacientes estudiados no se encontraron estas manifestaciones.
Los estudios para el diagnóstico incluyen la sideremia como marcador indirecto, que debe ser realizada en dos ocasiones. Especial valor tiene el IS mayor del 45 % y la ferritina sérica elevada. Este último es un excelente marcador pronóstico para predecir la fibrosis y la cirrosis cuando está por encima de 1 000 ng/mL. Sin embargo, no debe ser empleada como único marcador diagnóstico, ya que puede aumentar en otras enfermedades. La CT disminuida puede ser usada también como marcador de forma inversa al IS.1,3,10,15
En los enfermos homocigóticos para la C282Y, además de la elevación de la ferritina y las aminotransferasas, la presencia de diabetes, el consumo de alcohol y los bajos conteos de plaquetas son predictores de fibrosis, mientras que valores de IS inferiores al 45 % y ferritina normal pueden considerarse excluyentes para la enfermedad hereditaria por sobrecarga de hierro.3,15-17
De los pacientes estudiados, homocigóticos para la C282Y, solo en uno (No 2) se encontró diabetes y cirrosis, esta última comprobada por biopsia hepática, que apareció poco tiempo después del diagnóstico molecular. En este caso, aunque en el momento del estudio en el IHI la FS no se encontraba por encima de 1000 ng/mL, hay que considerar que el paciente había sido sometido a múltiples flebotomías anteriores (refirió más de 40).
En el paciente 1, también positivo para la C282Y, con FS y aminotransferasas elevadas, además de conteos de plaquetas subnormales desde el diagnóstico, los estudios imaginológicos realizados recientemente (4 años después) apuntan hacia una cirrosis, lo que reafirma el comportamiento habitual de la enfermedad.
En la era de la genética y la biología molecular, la determinación de las mutaciones más frecuentes (C282Y y H63D) constituye un pilar en el diagnóstico ante la sospecha de HH para definir el subtipo. Un resultado negativo para estas debería ir seguido del estudio de las otras mutaciones causantes de HH, sobre todo de los genes de hemojuvelina y hepcidina, por las graves implicaciones clínicas con que pueden cursar y su aparición en edades más tempranas de la vida. El monitoreo del resto de las mutaciones a través de secuenciaciones directas se reserva para investigaciones y casos con complicaciones severas, por lo que el tratamiento adecuado no debe depender del estudio molecular.6,10,17
La flebotomía o sangría ha sido empleada por más de 50 años para remover el exceso de hierro y aunque no hay datos que demuestren su influencia definitiva en la sobrevida global de los pacientes con HH por C282Y, es indudable su participación en la mejoría de los síntomas provocados por el exceso del mineral y por tanto, en la calidad de vida.5,17 El beneficio de la flebotomía está dado por la disminución del hierro circulante y la estimulación de la eritropoyesis que moviliza el hierro de los depósitos.
Con cada mL de eritrocitos extraídos se remueve 1 mg de hierro, lo que equivale a remover aproximadamente 200 mg del mineral de la circulación por cada 500 mL de sangre total. Se plantea que cuando se extraen 8 mg de hierro, la FS disminuye 1 ng/mL, lo que significa una reducción de alrededor de 25 ng/mL en cada extracción de 500 mL.3,17 Habitualmente, en una flebotomía el volumen a extraer oscila entre 200 a 500 mL de sangre total.
En los pacientes estudiados, así como en los que aún no tienen realizado el estudio molecular, la flebotomía ha constituido la base del tratamiento y para evitar la hipovolemia transitoria que puede presentarse como evento adverso, se realiza la reposición con solución salina fisiológica en igual volumen al extraído, con lo que se han obtenido resultados satisfactorios.
Otros aspectos que se monitorean exhaustivamente son la cifra de hemoglobina y el nivel de hierro, para evitar la anemia iatrogénica y la deficiencia del mineral. Es de destacar que en nuestra serie, a los 5 meses de mantener las flebotomías semanales el índice de saturación alcanzó una media del 46 %. Sin embargo, no sucedió igual con los niveles de FS, los que requirieron alrededor de 9 meses para alcanzar una media de 296,3 ng/mL. Esto pudiera estar relacionado con que el IS es una variable que depende del hierro circulante y este puede estar influenciado por la ingesta. En todos los casos, los pacientes refirieron mejoría de las manifestaciones clínicas después del tercer mes de tratamiento y ninguno ha vuelto a tener valores de FS por encima de 500 ng/mL.
La efectividad demostrada, la experiencia acumulada en su uso, la disponibilidad, seguridad y el bajo costo, unido a la posibilidad de mejorar la cirrosis, hacen de esta técnica el pilar principal del tratamiento en la HH. Sin embargo, HH molecular no es condición indiscutible y única para iniciarla, pues el 50 % de los individuos homocigóticos para la C282Y tienen FS normal y una menor proporción no tiene incremento progresivo de este marcador. Tampoco es un indicador de realizar flebotomía la sola elevación de las aminotransferasas.10,15,17
La eritrocitaféresis remueve mayor cantidad de hierro, menos proteínas plasmáticas, factores de la coagulación y plaquetas que la flebotomía, pero existen unas serie de factores que limitan su uso.1,17,18
En relación con el régimen higiénico-dietético, algunos clínicos prescriben una dieta pobre en hierro, pero esto no es totalmente adecuado porque influye negativamente en el estilo y calidad de vida de los enfermos.5,17 Sin embargo, algunas recomendaciones deben ser tenidas en cuenta: a) evitar la suplementación con hierro; b) consumo moderado de carnes rojas; c) ingestión de alcohol < 60 g/día; c) no consumir mariscos crudos, ni alimentos cocinados con agua de mar; d) consumir té negro y frutas frescas, preferiblemente no cítricas (2-3 veces/día) y limitar el consumo de vitamina C.1, 17 Algunos autores han recomendado tomar vitamina C como quelante. Sin embargo, esto puede aumentar la liberación de radicales libres por lo que hay que ser prudentes en su ingesta (no más de 500 mg/día).1,9,17
Los quelantes del hierro serian la alternativa terapéutica ideal para la HH y han mostrado ser eficaces en el tratamiento de la hemocromatosis secundaria.10,18 Diferentes quelantes, como la deferoxamina, la deferiprona y el deferasiroux se emplean en el tratamiento del exceso de hierro.1,3,5,17,19
Los inhibidores de la bomba de protones inhiben la absorción del hierro no hemo proveniente de la dieta, por lo que se han empleado en pacientes con HH.17
Finalmente, los agonistas de la hepcidina, agentes que remplazan la actividad de esta proteína o estimulan su producción endógena, pudieran ser muy útiles en la prevención de la acumulación del hierro. 20-21
La fisiopatología y diagnóstico de la HH ha avanzado notablemente en los últimos años. Sin embargo, el diagnóstico continúa realizándose de forma tardía y hacerlo en la fase presintomática contribuiría a evitar complicaciones.22 Ahora se imponen, por una parte, estudios que demuestren la efectividad y la influencia de la prevención en la sobrevida de los pacientes sin manifestaciones clínicas de la enfermedad; y por otra, la investigación de alternativas terapéuticas que pudieran incluir el uso de hepcidina exógena y sus análogos o agonistas.
REFERENCIAS BIBLIOGRÁFICAS
1. Siddique A, Kowdley KV. Review article: the iron overloads syndromes. Aliment Pharmacol Ther. 2012; 35:876-93.
2. Robson KJH, MerryweatherClarke AT, Cadet E, Viprakasit V, Zaahl MG, Pointon JJ et al. Recent advances in understanding haemochromatosis: a transition state. J Med Genet. 2004; 41:721-30.
3. Muñoz M, García-Erce JA, Remacha AF. Disorders of iron metabolismo. Part II: iron deficiency and iron overload. J Clin Pathol. 2011; 64:287-96.
4. Siah CW, Ombiga J, Adams LA, Trinder D, Olynyk JK. Normal iron metabolism and the pathophysiology of iron disorders. Clin Biochem Rev. 2006 Feb;27(1):5-16.
5. Beutler E, Hoffbrand AV, Cook JD. Iron deficiency and overload. Hematology Am Soc Hematol Educ Program. 2003:40-61.
6. Paulo CJ, Santo JL, Krieger JE, Pereira AC. Molecular diagnostic and pathogenesis of Hereditary Hemochromatosis. Int J Mol Sci. 2012; 13(2):1497-1511.
7. De Lima Santos PCJ, Luana-Dinardo C, Delfini Cançado R, Tadeu-Schettert I, Krieger JE, Costa-Pereira A. Non HFE Hemochromatosis. Rev Bras Hematol Hemoter. 2012;34(4): 311-16.
8. Sham RL, Phatak PD, Nemeth E, Ganz T. Hereditary Hemochromatosis due to resistance to hepcidin: high hepcidin concentrations in a family with C326S ferroportin mutation. Blood. 2009; 114:493-94.
9. Brissot P, BérengèreTroadec M, Bordon-Jacquet E, Le Lanc C, Jovanolle AM, Deugner I et al. Current approach to hemochromatosis. Blood Reviews. 2008; 22:195-210.
10. Fleming RE, Punka P. Iron overload in human disease. N Engl J Med. 2012; 366:348-59.
11. Finberg KE. Unraveling mechanisms regulating systemic iron homeostasis. Hematology Am Soc Hematol Educ Program. 2011;2011:532-7.
12. Forrellat-Barrios M, Fernández-Delgado N, Hernández-Ramírez P. Nuevos conocimientos sobre el metabolismo del hierro Rev Cubana Hematol Inmunol Hemoter. 2005 [consultado: 20 de agosto de 2012];.21(3): Disponible en: http://www.bvs.sld.cu/revistas/hih/vol21_3_05 /hih03305.htm
13. Forrellat-Barrios M, Fernández-Delgado N, Hernández-Ramírez P. Regulación de la hepcidina y homeostasia del hierro. Avances y perspectivas. Rev Cubana Hematol Inmunol Hemoter.2012 [consultado: 20 de agosto de 2012]; 28 (4): Disponible en: http://revhematologia.sld.cu/index.php/hih/article/view/3/9
14. Von Drygalski A, Pharm D, Adamson JW. Iron metabolism in man. J Parenteral Enteral Nutrition.2012; 20(10):1-8.
15. EASL clinical practice guidelines for HFE Hereditary Hemochromatosis. J Hepatol. 2010; 53(1):3-22.
16. Wood MJ, Powell LW, Dixon JL, Ramm GA. Clinical cofactors and hepatic fibrosis in Hereditary Hemochromatosis. The role of diabetes mellitus. Hepatology 2012; 56(3):904-11.
17. Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010:116:317-25.
18. Rombout-Sestrienkova E, von Noords PA, Von Deursen CT, Sybesma BJ, Nilleson- Meertens AE, Koeo GH. Therapeutic erythrocytapheresis versus phlebotomy in the initial treatment of Hereditary Hemochromatosis, A pilot study. Transfus Apher Sci. 2007; 36(3):261-7.
19. Cohen AR. New advances in iron chelation therapy. Hematology Am Soc Hematol Educ Program. 2006:42-7.
20. Pietrangelo A. Hepcidina in human iron disorders: Therapeutic implication. J Hepatology. 2011;54: 173-81
21. Ganz T, Nemeth E. The hepcidin and iron homeostasis. Biochemic Biophys Acta. 2012; 1823 (9):1434-43.
22. Cervera-García IA. Hemocromatosis tipo I. Patogenia y diagnóstico. Medisur.2012;10 (2):128-35.
Recibido: Febrero
20, 2013
Aceptado:
Junio 3,2013
Dra. Norma D. Fernández Delgado. Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA. Tel (537) 643 8695, 8268. Fax (537) 644 2334. Correo electrónico: rchematologia@infomed.sld.cu
{kind=link}