{kind=link}
{kind=link}
ARTÍCULO ORIGINAL
Resultados del tratamiento de la Leucemia Linfoide Aguda del niño en Cuba
Results of the treatment of child Acute Lymphoid Leukemia in Cuba
Dr. Alejandro González-OteroI, Dra. Andrea Menéndez-VeitíaI, Dr. Sergio Machín-GarcíaI, DraC. Prof. Eva SvarchI, Dra. Mirta Campo-DíazII, Dra. Raquel Fernández-NodarseIII, Dra. Liliana Martínez-CárdenasIV, Dra. Valia Pavón-MoránI, Dr. Esteban Márquez-HernándezV, Dra. Sonia Pérez-GarcíaVI, Dra. Rosa María Lam-DíazI, Dr. Alberto Arencibia-NúñezI, Dr. Juan Carlos Jaime-FagundoI, DraC. Consuelo Macías-AbrahamI, DraC. Ana María Amor-VigilI, Dra. Vianed Marsán-SuárezI
I Instituto
de Hematología e Inmunología, La Habana, Cuba.
II
Hospital Pediátrico "Pepe Portilla", Pinar del Río,
Cuba.
III
Hospital Pediátrico "Juan M. Márquez", La Habana, Cuba.
IV Hospital
Pediátrico "José Luis Miranda", Villa Clara, Cuba.
V
Hospital Pediátrico de Centro Habana, La Habana, Cuba.
VI
Hospital Pediátrico "Paquito González", Cienfuegos,
Cuba.
RESUMEN
Introducción:
la leucemia linfoide aguda (LLA) es la enfermedad maligna más frecuente
en la infancia y la primera que se trató con un protocolo común
en Cuba. Se han aplicado diferentes protocolos a lo largo del tiempo y actualmente
existen en el país 10 centros que tratan niños con LLA.
Objetivo:
presentar los resultados alcanzados en el tratamiento de la LLA desde 2002 hasta
2008 con el protocolo del grupo ALLIC (Acute Lymphoblastic Leukemia Intercontinental).
Métodos: se trataron 166 niños menores de 18 años al
inicio de la enfermedad. Para conformar los grupos pronóstico se utilizaron
diferentes criterios que incluyeron la edad y el número de leucocitos
en el momento del diagnóstico, las alteraciones moleculares y la respuesta
al tratamiento.
Resultados:
la supervivencia libre de eventos (SLE) a los 4 años fue del 69 %
y la supervivencia global del 78 %. La SLE en los diferentes grupos pronósticos
fue del 85 % para los pacientes de riesgo estándar, 77 % para los de
riesgo intermedio y 59 % para los de riesgo alto. El porcentaje de remisión
inicial fue inferior al obtenido por el grupo total. La mayoría de las
muertes ocurrieron al inicio de la aplicación del protocolo. La recaída
hematológica fue la causa más frecuente de terminación
de la remisión completa. Las recaídas del sistema nervioso central,
las testiculares y las combinadas fueron inferiores al 5 %. La presencia de
reordenamientos genéticos del tipo bcr/abl o MLL/AF4 se confirmaron como
elementos de muy mal pronóstico.
Conclusiones:
estos resultados, aunque son susceptibles de ser mejorados, muestran
un nivel adecuado, sobre todo si se tiene en cuenta que se han logrado en un
país en vías de desarrollo.
Palabras clave: leucemia linfoide aguda pediátrica, grupos pronóstico, supervivencia global, supervivencia libre de eventos.
ABSTRACT
Introduction:
Acute lymphoid leukaemia (ALL) is the most frequent malignant disease in
childhood and the first one to be treated with a common protocol in Cuba.
Different protocols have been used and at present there are 10 health centers
in Cuba treating children with ALL.
Objective:
To present the results achieved in the treatment of ALL from 2002 to 2008 with
protocol ALLIC (Acute Lymphoblastic Leukemia Intercontinental).
Methods:
166 children under 18 years old at the beginning of the disease were treated.
Different criteria were used to make prognostic groups which included age and
leukocyte counts in the peripheral smear at diagnosis, DNA molecular rearrangements
and response to therapy.
Results:
Event free survival (EFS) after 4 years for the whole group was 69 % and overall
survival (SV) was 78 %. EFS in the different prognostic groups were 85 % for
standard risk patients, 77 % for the intermediate risk group and 59 % for high
risk children. Percentage of initial remission in our patients was lower than
the one obtained for the whole group. The majority of early deaths occurred
at the beginning of the protocol application. Bone marrow relapses were the
more frequent ones. Central nervous system, testicular or combined relapses
were lower than 5 %. DNA rearrangements for bcr/ abl or MLL/AF4 were signs of
very bad prognosis.
Conclusions:
These results, even when susceptible to be better, show an adequate level
considering that they were achieved in a developing country.
Key words: Acute lymphoblastic leukemia, prognostic groups, overall survival, event free survival.
INTRODUCCIÓN
El tratamiento de las leucemias agudas en la edad pediátrica comenzó a realizarse en Cuba con protocolos establecidos, a partir del año 1973. La primera enfermedad que se trató con un protocolo común fue la leucemia linfoide aguda (LLA) que es la enfermedad maligna más frecuente en la infancia (80 % de todas las leucemias en el niño) y no se comenzó a realizar un tratamiento uniforme en todo el país hasta 1986 en que se formó el Grupo de Estudio y Tratamiento de Hemopatías Malignas en Cuba (GETHMAC). Desde el inicio hasta el año 2002 se trataron más de 900 niños menores de 18 años. Se utilizaron diferentes protocolos, con resultados que fueron mejorando progresivamente a medida que se fueron aplicando esquemas más avanzados.1-3 A partir del año 1982 se incorporaron los protocolos basados en los esquemas terapéuticos del grupo alemán Berlín-Frankfurt-Munster (BFM), que contribuyeron a mejorar los resultados obtenidos hasta entonces.4,5
En los últimos 30 años ocurrieron numerosos avances en el conocimiento de la biología de la enfermedad que han permitido el establecimiento de factores pronósticos,6 tanto clínicos como citomorfológicos, mediante la clasificación del grupo Franco Americano Británico (FAB);7 además de la caracterización inmunofenotípica 8-10 y el estudio de las alteraciones citogenéticas11 y moleculares.12 El conocimiento de la mayor cantidad de factores pronósticos desde el momento del diagnóstico permite, en la mayoría de los casos, predecir la evolución de la enfermedad y ofrece la posibilidad de modificar el tratamiento en los pacientes de alto riesgo.
A partir del año 2002 se estableció el grupo cooperativo ALLIC (siglas del inglés, Acute Lymphoblastic Leukemia Intercontinental) , que emplea un protocolo terapéutico de tipo BFM sin detección de la enfermedad mínima residual (EMR) y en el que participan más de 20 países de diferentes continentes. Cuba aplica este protocolo para el tratamiento de la LLA del niño. En el país existen 10 centros que tratan niños con LLA pero en algunos no es posible realizar los estudios moleculares ni del inmunofenotipo.
El objetivo de este trabajo fue describir los resultados obtenidos en los últimos años en el tratamiento de la LLA del niño en la mayoría de los centros del país.
MÉTODOS
Se incluyeron 166 pacientes menores de 18 años de edad que fueron diagnosticados entre noviembre de 2002 y octubre de 2008. Ochenta y cuatro (50,6 %) fueron atendidos en el Instituto de Hematología e Inmunología (IHI), que es el centro coordinador de la especialidad en el país.
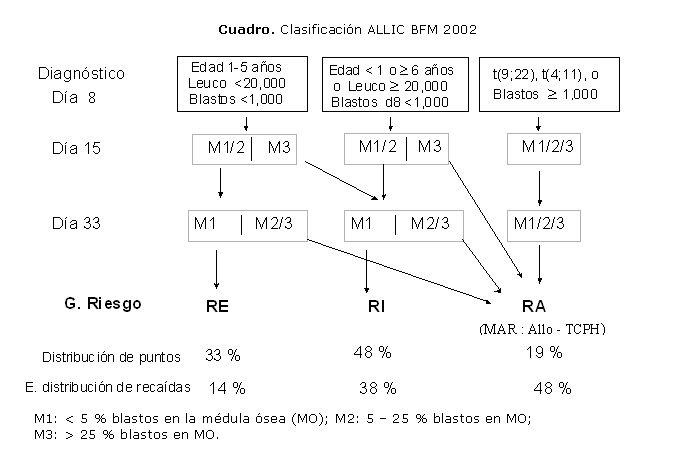
Los pacientes se
clasificaron según los criterios ALICC-BFM 2002 en riesgo estándar
(RE); riesgo intermedio (RI); riesgo alto (RA); además, se definió
un grupo de muy alto riesgo (MAR).
Esta clasificación incluye la presencia de las translocaciones t(9,22),
t(4,11) o el reordenamiento de los oncogenes bcr/abl y MLL/AF4 como elementos
de alto riesgo. Los estudios de inmunofenotipaje y de biología molecular
para todos los pacientes incluidos se realizaron en los laboratorios respectivos
del IHI con las técnicas habituales9-12 (Cuadro).
El tratamiento en general fue similar al protocolo BFM 200013 y las fases fundamentales y los principales medicamentos utilizados se exponen a continuación:
Inducción
Esta fase constó de dos etapas: el protocolo Ia con prednisona (P) oral por 29 días disminuyendo en una semana, vincristina (VCR) y daunomicina (DNM) una dosis semanal endovenosa (ev), excepto en los pacientes de RE que solo recibieron dos dosis de DNM y L-asparaginasa (L-Asa) 8 dosis, que se administraron por vía intramuscular (IM), nunca coincidiendo con la VCR. La profilaxis de la leucemia meníngea se realizó con tratamiento intratecal (it) con metotrexato (MTX) en tres ocasiones.
La segunda parte o protocolo Ib se aplicó a todos los pacientes con ciclofosfamida (CFM) ev al inicio y al final y cuatro ciclos de citosina arabinósido (Ara-C) de 4 días cada uno con 6 mercaptopurina (6MP) durante toda la fase, y dos tratamientos it con MTX.
Consolidación
La consolidación fue diferente para los enfermos de RA. Los de RE y RI inicialmente recibieron el llamado protocolo M con 4 administraciones de dosis elevadas de MTX ev (2 g/m2) cada 15 días, acompañado con la administración it de MTX y 6MP durante todo el tiempo, a los pacientes con inmunofenotipo T se les administró el MTX ev en una dosis de 3 g/m2. A continuación se efectuó una selección al azar por una tabla de números aleatorios para definir el tipo de consolidación tardía con protocolos II o III. El protocolo II tiene dos etapas: la primera o IIa con dexametasona (DXM), VCR y adriamicina (DOX) semanalmente, y cuatro dosis de L-Asa; en la segunda parte o IIb recibieron una dosis de CFM y dos ciclos de Ara-C de cuatro días cada uno con dos tratamientos it y 6MP. En el protocolo III solo recibieron dos dosis de VCR y DOX sin CFM en la segunda parte del tratamiento, aunque el resto fue similar.
Los pacientes de RE recibieron un protocolo II o dos protocolos III; y los pacientes de RI recibieron un protocolo II o tres protocolos III.
A los enfermos considerados de RA, después de la etapa de inducción con las dos partes del protocolo I se les administraron tres bloques de quimioterapìa intensa:
- Bloque RA-1: Se emplearon DMT, MTX y Ara-C en dosis altas, VCR, CFM y dosis altas de L-Asa. Tratamiento IT con MTX, Ara-C y DXM, en dos ocasiones.
- Bloque RA-2: Se utilizó DXM, VCR, DNR, dosis altas de MTX, Ifosfamida (IFO) y dos dosis de L-Asa con tratamiento It, similar al bloque anterior.
- Bloque RA-3: DEX, dosis altas de Ara-C, etopósido (VP-16) y dos dosis de L-Asa con tratamiento it similar a los dos bloques anteriores.
La duración de cada uno de los bloques fue de once días con administración de factor estimulador de colonias granulocíticas hasta la recuperación hematológica.
Posteriormente se aplicaron dos protocolos II, previo al mantenimiento. En el protocolo están contempladas otras variantes de tratamiento para este grupo de pacientes, pero debido a que el número de enfermos esperado no era muy grande, el grupo internacional decidió que cada país escogiera una variante sin selección al azar.
Se utilizó radioterapia craneal profiláctica a 12 Gy solo en los pacientes con inmunofenotipo T y en todos los de RA. Los pacientes con infiltración meníngea inicial recibieron 18 Gy.
Mantenimiento
La terapia de mantenimiento se realizó con 6MP diaria y MTX una vez a la semana. La duración varió en cada grupo de riesgo de acuerdo con la duración de la etapa de consolidación; en los pacientes de RE y RI se hizo un tratamiento IT con MTX adicional mensual en los primeros cuatro meses. La duración total del tratamiento para todos los grupos de riesgo fue de 104 semanas, independientemente del sexo.
La información de los pacientes se registró en una base de datos que se procesó con el programa SSPS v 11.5. La sobrevida libre de eventos (SLE) y la sobrevida global (SG) a los 60 meses se determinaron por el método de Kaplan Meier. Las diferencias entre las curvas se analizaron por la prueba de log rank. Cuando hubo entrecruzamiento entre las curvas se aplicó el test de Breslow para analizar la significación estadística. El nivel de significación escogido fue de p<0,05.
RESULTADOS
En el período de ejecución del protocolo de tratamiento de la LLA desde noviembre del año 2002 hasta el 31 de octubre de 2008, fecha de terminación del estudio, se incluyeron 166 pacientes menores de 18 años procedentes de 6 centros hospitalarios del país.
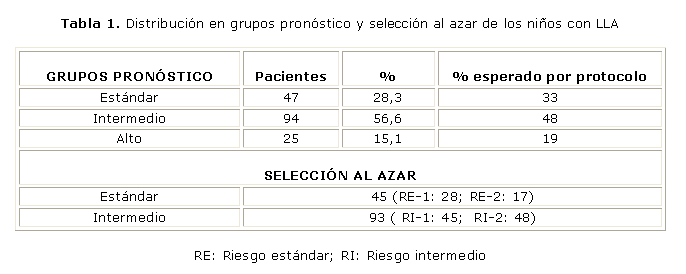
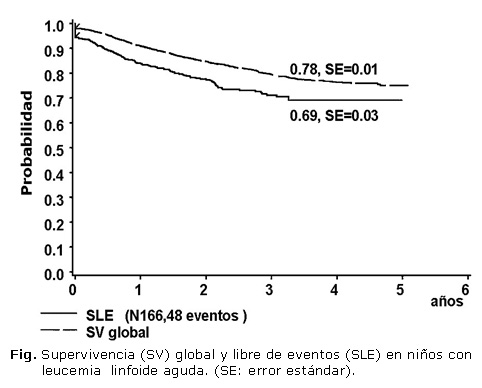
Se trataron 101 pacientes del sexo masculino (60,8 %) y 65 del femenino (39,2 %). La distribución por grupos pronóstico y la selección al azar se muestran en la tabla 1. La SLE y la SG del total de los pacientes fueron del 69 y 78 % respectivamente. (Figura). No hubo diferencias en estas curvas entre los pacientes que recibieron protocolo II o protocolo III como consolidación tardía, pero la toxicidad fue mayor en este último grupo. La SLE según los grupos pronóstico fue superior al 80 % para los de RE y menor del 60 % para los de RA.
Se pudo realizar el inmunofenotipo en 119 pacientes y no fue posible efectuar los estudios en 47, debido a que no se obtuvieron células o a que no se enviaron las muestras desde otros centros. Hubo 96 pacientes con inmunofenotipo B (80,66 %), 19 con marcadores T (15,9 %) y 4 con inmunofenotipo híbrido (3,36 %). Los estudios de biología molecular por PCR se realizaron en 115 enfermos; en 6 (5,21 %) se encontró reordenamiento bcr/abl, en 2 (1,7 %) reordenamiento del gen MLL, y en 22 (19,1 %) reordenamiento TEL7AML1.
Al terminar la primera parte del tratamiento de inducción 150 pacientes (90,4 %) alcanzaron la remisión inicial. Siete niños murieron durante la inducción por infecciones o hemorragias y en 9 pacientes no se obtuvo la remisión debido a que tuvieron una enfermedad resistente. El porcentaje de remisión inicial varióen los diferentes centros desde 94 hasta 76 % y fue del 97 % para el grupo de RE, 93 % para el RI y 64 % para el RA.
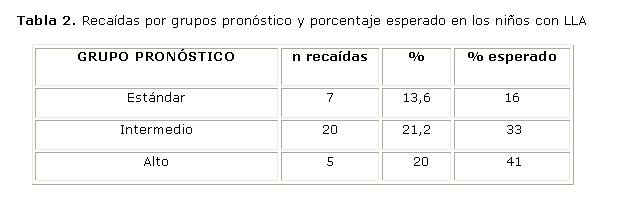
Se produjeron 32 recaídas, de las que 22 (68,8 %) fueron en la médula ósea, 5 (15,6 %) aisladas en el SNC y 5 (15,6 %) fueron combinadas en la médula ósea y el SNC, o en la médula ósea y el testículo. Cuando se analizó el porcentaje en relación con el total de pacientes incluidos, la recaída hematológica ocurrió en el 14,6 % de los enfermos estudiados y la del SNC en el 3,3 %. Se diagnosticaron más recaídas en el grupo de RA (tabla 2).
Cuatro pacientes fallecieron en remisión y 15, en recaída. De los seis pacientes con reordenamiento bcr/abl, cromosoma de Filadelfia positivo (Ph+), 5 recayeron y fallecieron, al igual que los dos con reordenamiento del gen MLL. Solo se realizó un trasplante alogénico de células progenitoras hematopoyéticas (TCPH) en un paciente con una LLA Ph+, que falleció en el período postrasplante inmediato.
DISCUSION
Los resultados obtenidos en el tratamiento de la LLA con el protocolo ALLIC 2002 fueron mejores que los alcanzados previamente en el país, pero inferiores a los descritos por el grupo ALLIC en su totalidad, que obtuvo una SLE del 76 % a los 4 años.13
El porcentaje de remisión inicial fue inferior a lo logrado por la mayoría de los grupos cooperativos que describen remisiones hematológicas iniciales superiores al 95 %.14-16 Hubo diferencias entre los distintos centros del país, ya que en algunos de los hospitales participantes la remisión inicial fue inferior al 80 %. Es probable que estos resultados se deban a la necesidad de un tratamiento de soporte más eficaz y a mejores condiciones de aislamiento en algunos centros.
Dentro del grupo BFM se ha señalado que tanto el tiempo de experiencia en el manejo de los protocolos como el número total de pacientes tratados por año, que nunca debe ser inferior a 10, influyen en la posibilidad de alcanzar mejores resultados iniciales.17 En nuestro grupo la mayoría de las muertes en inducción ocurrieron en la etapa inicial de aplicación del protocolo.
Un aspecto interesante en este trabajo es que el número de pacientes en los diferentes grupos de riesgo no correspondió con lo que se esperaba de acuerdo con el diseño estadístico del protocolo. Al respecto es posible que la dificultad para realizar los estudios citogenéticos y moleculares a todos los enfermos haya influido en que se considerara un mayor número de pacientes como RI; sin embargo, este aspecto no repercutió en el porcentaje de recaídas esperadas. En general, los resultados del inmunofenotipo coinciden con lo descrito en la literatura. Hay que señalar que la evolución de los pacientes con reordenamiento bcr/abl es desfavorable en los centros de países desarrollados.18 Nuestros resultados coinciden con estos hallazgos aunque la muestra del presente estudio fue pequeña.
En la actualidad, con la administración del Imatinib o de otros inhibidores de la tirosina cinasa se han obtenido mejores resultados en algunos pacientes con este reordenamiento.19-22
Solo fue posible realizar un TCPH por las características de la población cubana en la que es muy difícil obtener donantes relacionados debido a que la mayoría de las familias sólo tienen uno o dos hijos. Además, la composición étnica determinada por la mezcla de genes europeos y africanos hace poco probable obtener donantes no relacionados en los bancos de células progenitoras hematopoyéticas.
La SLE y la SV global del grupo fueron aceptables aunque, como se señaló anteriormente, la posibilidad de TCPH es muy limitada en Cuba, al igual que otros tratamientos de rescate en pacientes con recaídas no muy precoces. La SLE en los diferentes grupos pronóstico fue similar a la descrita en la literatura, lo que demuestra la utilidad del sistema de clasificación empleado, y aunque aún el tiempo de seguimiento no ha permitido que se alcance una meseta, se observaron diferencias significativas entre los niños clasificados como RE y RI con los de RA.
Aún cuando nuestros resultados son favorables es necesario realizar varias acciones y modificaciones a la situación actual. Uno de los aspectos imprescindibles es la incorporación de todos los pacientes diagnosticados con LLA en el país para realizar todos los estudios de clasificación 23,24 y de esa manera poder definir el tratamiento más adecuado.
El grupo cooperativo ALLIC comenzó un nuevo estudio que incluye la detección de la EMR por citometría de flujo el día 15 de tratamiento.25,26 Realizar los estudios de inmunofenotipaje en este momento ofrece una información de gran valor sin tener que estudiar la EMR en otras oportunidades, con el consiguiente ahorro de recursos que este enfoque representa.27-31 Otra de las ventajas del estudio de la EMR sería poder realizar un tratamiento más específico a cada paciente para evitar, en lo posible, la ocurrencia de secuelas, en ocasiones relevantes.
Es importante insistir en la necesidad de tener servicios de hematología con condiciones de aislamiento adecuadas, un tratamiento de sostén eficaz y un equipo médico adiestrado, para obtener buenos resultados en instituciones de países con escasos recursos económicos.
Es necesario destacar la contribución del IHI para lograr los resultados obtenidos pues, como en todos los tipos de leucemias agudas de la infancia, ha tenido no solo el carácter rector de la actividad en el país sino que ha atendido a la mayor cantidad de pacientes.
Los resultados en el tratamiento de la LLA en la edad pediátrica presentados, aunque son susceptibles de ser mejorados, muestran un nivel adecuado, sobre todo si se tiene en cuenta que se han logrado en un país en vías de desarrollo.
AGRADECIMIENTOS
A colegas de la rama pediátrica del Grupo de Estudio y Tratamiento de Hemopatías Malignas en Cuba (GETHMAC), por su valiosa colaboración en el suministro de información imprescindible para la elaboración de este trabajo: doctores Ileana Nordet, Adis Gutiérrez, Jesús Serrano, Aramís Núñez y Marlén Domínguez (Instituto de Hematología e Inmunología, La Habana); Edeli Rosell, Loreta Peón, Orestes Chágues e Ileana Ortiz (Hospital Pediátrico "Juan M. Márquez", La Habana); Jorge Luis Hernández (Hospital Pediátrico "Pepe Portilla", Pinar del Río); y Bertha Vergara y Tamara Cedré (Hospital Pediátrico "José Luis Miranda", Villa Clara)
Igualmente, nuestro reconocimiento a otros hematólogos pediatras que han atendido a estos pacientes en los distintos centros hospitalarios cubanos.
REFERENCIAS BIBLIOGRÁFICAS
1. Svarch E, González A, Lagarde M, Vergara B. Tratamiento de la LLA del niño. Bol Med Hosp Infantil México.1980: 37:707-14.
2. Svarch E, Vergara B, Lagarde M, González A, Quintero I, Fernández O. et al. Tratamiento de la leucemia linfoblástica aguda (LLA) Sangre.1980;25:411-6.
3. Svarch E, Vergara B, González A, Lagarde M. Tratamiento de la leucemia linfocítica aguda. Rev Cubana Invest Biomed.1982;1(1):38-44.
4. González A, Vergara B, Svarch E. Resultados preliminares del tratamiento de la leucemia aguda linfoblástica (LAL) en Cuba con los protocolos 3-LLA-82 y 1-LLA-84 Rev Cubana Hemat Inmunol Hemoter. 1986; 21: 263-8.
5. Svarch E, González A, Vergara B, Campo M, Méndez J, Fernández O, et al. Tratamiento de la leucemia linfoide aguda (LLA) en el niño. Sangre.1993; 38(1): 251-7.
6. González A, Pérez A, Svarch E. Valor de la clasificación en grupos pronósticos de la leucemia aguda linfoblástica de la infancia Rev Cubana Hemat Inmunol Hemoter.1989; 5(3):436-40.
7. Bennett JC, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, et al. Proposed revised criteria for the classification of acute myeloid leukemia. A report of the French-American-British Cooperative Group. Ann Intern Med.1985 Oct;103(4):620-5.
8. Pui CH, Relling MV, Pharm D, Downing JR. Acute lymphoblastic leukemia 2004 Apr;350(15):1535-48.
9. Campana D, Coustan-Smith F. Advances in the immunological monitoring of childhood acute lymphoblastic leukaemia. Best Pract Res Clin Haematol2002 Mar;15(1):1-19.
10. Marsán-Suárez V, del Valle-Pérez L, Socarrás-Ferrer B, Sánchez-Segura M, Macías-Abraham C, Mazorra-Herrrera Z, et al Validación del ultramicrométodo inmunocitoquímico (UMICIQ) para el inmunofenotipaje de la leucemia linfoide pediátrica Rev Cubana Hematol Inmunol Hemoter. 2012 Abr-Jun;29 (3) 74-78.
11. Jakovljovic G, Nakic M, Rogosic S, Kardum-Skclic I, Zadro R, Kroslic B. Pre-B-cell acute lymphoblastic leukemia with bulk extramedullary disease and Chromosome 22 (EWSR1) rearrangement masquerading as Ewing sarcoma. Pediatr Blood Cancer.2010 Apr;54(4):606-9.
12. Pui CH, Evans WE. Acute lymphoblastic leukaemia. N Engl J Med. 1998 Aug;339(9):605-15.
13. Armstrong SA, Look AT. Molecular genetics of acute lymphoblastic leukaemia. J Clin Oncol. 2005 Sep;23(26):6306-15.
14. Pui CH. Childhood leukemias. N Engl J Med. 1995 Jun;332(24):1618-30.
15. Assumpção JG, Ganazza MA, de Araújo M, Silva AS, Scrideli CA, Brandalise SR, etal. Detection of clonal Immunoglobulin and T-cell receptor gene rearrangement in childhood acute lymphoblastic leukemia using a low-cost PCR strategy. Pediatr Blood Cancer. 2010 Dec;55(7):1278-86. doi:10.1002/pbc.22709.
16. Ritter J, Creutzig U, Reier A, Riehm H, Schellong G. Childhood leukemias: Cooperative Berlin-Frankfurt-Munster trials in Federal Republic of Germany. J Cancer Res Clin Oncol.1990;116(1):100-3.
17. ALLIC. ALLIC-BFM 2002 Protocol progress reports 23-30, 20th Annual Meeting of the International BFM Study Group May 8th to 10th 2009, Bergamo, Italy.
18. Duration and intensity of maintenance chemotherapy in acute lymphoblastic leukaemia: overview of 42 trials involving 12 000 randomized children. Childhood ALL Collaborative Group. Lancet.1996 Jun;347(9018):1783-8.
19. Verdiman HJ, Kamps WA, van en Berg H, van den Berg E, Bokkrink JPA, Bruin CA, et al. Dexamethasone based therapy for childhood acute lymphoblastic leukaemia results of the prospective Dutch Childhood Oncology Group (DCOG) protocol ALL-9 (1997-2004). Lancet Oncol. 2009 Oct;10(10):957-66. doi:10.1016/S1470-2045(09)70228-1.
20. Biondi A, Baruchel A, Hunger S, Masera G, Schmiiegelow K, Schrappe M, et al. The eleventh international childhood acute lymphoblastic leukemia workshop Ponte di Legno Italy 6-7 may 2009. Leukemia.2009 Dec;23(12):2318-24.
21. Yanada M, Takeuchi J, Sugiura I, Akiyama H, Usui N, Yagasaki F, et al. High complete remission rate and promisingoutcome for newly diagnosed BCR-ABL-positive acute lymphoblastic leukemia. A phase II study by the Japan Acute Leukemia Study Group. J Clin Oncol.2006 Jan;24(3):460-6.
22. Sazawal S, Bakhshi S, Raina J, Swaroop Csayen R. Detection and clinical relevance of bcr-abl fusion gene in T cell lineage ALL .Report of four cases: J Pediatr Hematol Oncol. 2009 Nov;31(11):850-2.
23. Vrooman LM, Silverman LB. Childhood acute lymphoblastic leukemia. Update of prognostic factors Curr Opin Pediatr. 2009:21(1) 1-8.
24. Pui CH. T cell lymphoblastic leukemia NOTCHing the way towards a better treatment outcome Cancer Cell 2009 Feb;15(2) 85-7. doi:10.1016/j.ccr.2009.01.007.
25. Gaipa G, Basso G, Maglia O, Leoni V, Faini A, Cazzaniga G, et al. Drug-induced immunophenotypic modulation in childhood ALL: Implications for minimal residual disease detection . Leukemia.2005 Jan;19(1):49-56.
26. Campana D. Status of minimal residual disease testing in childhood haematological malignancies. Br J Haematol.2008 Nov;143(4):481-9.
27. Burnett AK, Eden OB. The treatment of acute leukaemia. Lancet.1997 Jan;349(9047):270-5.
28. Fichr T, Schrauder A, Cazzaniga C, Panzer-Grümayer P, van der Velden V, Fischer S, et al. On behalf of the International BFM Study Group (I-BFM-SG) Minimal residual disease directed risk stratification using real-time quantitative PCR analysis of immunoglobulin and T cell receptor gene rearrangements in the international multicenter trial AIEOP-BFM ALL 2000 for childhood acute lymphoblastic leukemia. Leukemia2008 Apr;22(4):771-82.
29. Attarbaschi A, Marin G, Panzer-Grúmayer P, Rutgers S, Steiner M, Königen M, et al. Minimal residual disease values discriminate between low and high risk in children with B cell precursor acute lymphoblastic leukemia and intrachromosomal amplification +of chromosome 21: The Austrian and German acute Lymphoblastic leukemia Berlin-Frankfurt-Münster (ALL-BFM) trial. J Clin Oncol. 2008 Jun;26(18):3046-50.
30. Yamaji K, Okamoto T, Yokota S, Watanabe A, Horikoshi Y, Asami K, et al. Minimal residual disease based augmented therapy in childhood acute lymphoblastic leukemia: A report from the Japanese childhood cancer and leukemia study group. Pediatr Blood Cancer.2010 Dec;55(7):1287-95.
31. Basso G, Veltroni M, Valsecchi MG, Henze G, Ludwig WD, Schumich A, et al. Risk of relapse of childhood acute lymphoblastic leukemia is predicted by flow cytometric measurement of minimal residual disease on day 15 bone marrow. J Clin Oncol. 2009: 27 (31): 5168-74.
Recibido: Agosto
26, 2013.
Aceptado: Septiembre
12, 2013
Dr. Alejandro González Otero. Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA. Tel (537) 643 8695, 8268. Fax (537) 644 2334. Correo electrónico: rchematologia@infomed.sld.cu
{kind=link}