ARTÍCULO ORIGINAL
Policitemia Vera en la era del Jak: breve análisis del diagnóstico después de su introducción
Polycythemia Vera at Jak age: brief analysis of the diagnosis after its introduction
Dra Norma Fernández Delgado, Dr. Darwin Martínez Ferrufino, Dr. Roy Román Torres, DraC. Ana María Amor Vigil, Ing Teresa Fundora Sarraff, Dra Ivis Macía Pérez, Lic. Laser Hernández Reyes.
Instituto de Hematología e Inmunología. La Habana, Cuba.
RESUMEN
INTRODUCCIÓN:
la Policitemia Vera es una neoplasia mieloproliferativa BCR/ABL negativa en
la que se ha descrito la presencia de la mutación JAK2V617F en más
del 90 % de los afectados. En el Instituto de Hematología e Inmunología
se introdujo la determinación de esta mutación desde la segunda mitad
del 2008.
Objetivos: identificar el porcentaje de pacientes positivos para el JAK2V617F
y correlacionar esta con las variables posibles a estudiar en el país.
MÉTODOS: se realizó un estudio descriptivo que incluyó
45 pacientes, 15 con diagnóstico anterior y 30 de reciente diagnóstico.
Todos fueron evaluados según los criterios OMS 2001 y 2008, con las variables:
eritrocitosis demostrada, ausencia de causas secundarias, vísceromegalia,
resultados del hemograma, biopsia de médula ósea al diagnóstico
y estudio molecular.
RESULTADOS: La mutación resultó positiva en el 86,6 % (n =
39) de los pacientes en los que el diagnóstico se confirmó por ambos
criterios OMS. Dos pacientes fueron considerados neoplasias mieloproliferativas
no clasificadas. La edad fue menor en los negativos para la mutación (p
= 0.03). La presencia de la mutación se relacionó con cifras de leucocitos
ligeramente superiores, trombocitosis significativa (p = 0,000) y una histología
de la médula ósea con mayores cambios morfológicos.
CONCLUSIONES: Estos resultados muestran que es posible realizar el diagnóstico
de la PV con el empleo de los criterios de la OMS del 2001, aunque el estudio
molecular facilita la precisión diagnóstica y contribuye a reducir
el margen de error diagnóstico. Sin embargo, una revisión objetiva
de los criterios actuales podría garantizar un diagnóstico certero
en cualquier lugar del mundo.
Palabras clave: policitemia vera, mutación JAKV617F, criterios OMS
ABSTRACT
INTRODUCTION:
Polycythemia vera is a BCR/ABL negative myeloproliferative neoplasm in which
the JAK2V617F mutation has been described in more than 90 % of cases. At the
Institute of Hematology and Immunology determination of this mutation was introduced
since the second half of 2008.
OBJECTIVES: To identify the percentage of positive patients for the JAK2V617F
mutation and correlate this with the remaining variables possible to be studied
in Cuba.
METHODS: A descriptive study that included 45 patients, 15 previously
diagnosed and 30 of new diagnosis, was carried out. All patients were evaluated
according to 2001 and 2008 WHO criteria, with the following variables: demonstrated
erythrocytosis, absence of secondary causes, visceromegaly, results of hemogram,
bone marrow biopsy at diagnosis and molecular study.
RESULTS: Mutation was positive in 86,6% (n = 39) of the patients with
confirmed diagnosis by both WHO criteria. Two patients were considered unclassificable
myeloproliferative neoplasms. Age was smaller in patients negative for the mutation
(p = 0.03). The presence of the mutation was related to slightly superior leukocyte
count, significant thrombocytosis (p = 0,000) and larger morphological changes
in bone marrow histology.
CONCLUSIONS: These results show that it is possible to make the diagnosis
of PV with the use of 2001 WHO criteria, but the molecular testing leads to
an accurate diagnosis and contributes to reduce the rate of error. However,
an objective review of current criteria might guarantee an accurate diagnosis
in any place of the world.
Keyword: Polycythemia vera, JAK2V617F mutations, WHO criteria
INTRODUCCIÓN
La Policitemia vera (PV) es la más frecuente de las neoplasias mieloproliferativas crónicas (NMP) donde no hay presencia del cromosoma Filadelfia (Ph) o su gen de fusión BCR/ABL, y la causa más frecuente de eritrocitosis primaria. Se caracteriza por un incremento absoluto y autónomo de la masa eritrocitaria, trombocitosis, leucocitosis y esplenomegalia. Frecuentemente, su aparición es insidiosa y tiene una evolución progresiva que puede llegar a mielofibrosis o leucemia aguda.1-4
En el año 2005, diferentes grupos de investigadores describieron la presencia de la mutación JAK2V617F en el exón 14 del gen Janus Kinasa 2 del cromosoma, 9 en pacientes con NMP negativos para el Ph y su mayor positividad se ha comunicado en los granulocitos de pacientes con PV. Diferentes publicaciones citan su presencia entre el 65 y el 98 % de los pacientes estudiados.5,6 Posterior a ello, en el 2008, la Organización Mundial de la Salud (OMS) definió nuevos criterios diagnósticos para la PV que incluyen esta mutación entre los criterios mayores.7 Estos criterios incluyen, además de la presencia del JAK2V617F o de otra alteración molecular similar que evidencie clonalidad, demostrar la eritrocitosis, ya sea por cifras de hemoglobina (Hb) superiores a 185 g/L en el hombre y 165 g/L en la mujer, en varias determinaciones, u otra evidencia de incremento de la masa eritrocitaria (estudios de volúmenes sanguíneos). Los criterios menores incluyen la biopsia de médula ósea sugestiva de PV (hipercelularidad para la edad, prominente proliferación eritroide y panmielosis), la disminución de la eritropoyetina sérica y el crecimiento espontáneo de colonias eritroides in vitro. Para el diagnóstico son necesarios los dos criterios mayores y uno menor o la presencia de eritrocitosis con dos de los criterios menores.4,7-9
En Cuba y específicamente en el Instituto de Hematología e Inmunología (IHI), desde 2002 se emplean los criterios de la OMS del 2001 para el diagnóstico de la PV. Estos incluyen como criterios mayores demostrar la eritrocitosis, la ausencia de causas secundarias, la presencia de esplenomegalia, la evidencia de clonalidad no relacionada con el cromosoma Ph o del gen BCR/ABL, y el crecimiento espontáneo de colonias eritroides. En los criterios menores se relacionan la trombocitosis, la leucocitosis, la existencia de una biopsia de médula ósea sugestiva de la enfermedad y la disminución de la eritropoyetina sérica. Para el diagnóstico se requiere la presencia de tres criterios mayores o los dos primeros mayores con dos menores.8,9
Dadas las limitaciones para realizar algunos de los estudios propuestos en los criterios OMS del 2008 y teniendo en cuenta que la mutación del JAK2 no está presente en el 100 % de los pacientes, nos propusimos evaluar el valor de la mutación JAK2V617F en el diagnóstico de la PV en pacientes previamente diagnosticados y de nuevo diagnóstico, con el empleo de los criterios posibles de la OMS del 2001 a los que se les adicionó el estudio de la mutación del JAK.
MÉTODOS
Se realizó un estudio prospectivo, descriptivo de tipo transversal de pacientes provenientes de la consulta de Poliglobulias del IHI, en el período comprendido entre enero de 2010 y diciembre de 2012, que incluyó 45 pacientes: 15 con diagnóstico de PV anterior a la fecha, a los que se les realizó, además, el estudio molecular del JAK2V617F, y 30 de nuevo diagnóstico. A todos se les aplicaron los criterios diagnósticos de la OMS del 2001 y del 2008, atendiendo a las siguientes variables: eritrocitosis demostrada, ausencia de causas secundarias (ecocardiograma, estudios de función respiratoria, electroforesis de hemoglobina, ultrasonidos renal y abdominal), presencia de esplenomegalia palpable o por ultrasonografía, leucocitosis, trombocitosis, biopsia de médula ósea sugestiva de PV y estudio molecular para descartar la presencia del cromosoma Ph y evaluar la de la mutación JAK2V617F.
Todos los pacientes emitieron su consentimiento informado para la realización de los estudios moleculares y la utilización de los datos contenidos en su historia clínica.
La información se analizó por el método estadístico descriptivo y se utilizó el SPSS versión 15.0 para Windows. Para las variables cualitativas se utilizaron las frecuencias absolutas y los porcentajes; y para las cuantitativas, la media y la desviación estándar. Se empleó la prueba Chi Cuadrado de independencia para determinar asociación entre las variables con la presencia de la mutación. Se utilizó la prueba t Student para comparar los promedios de los resultados de laboratorio entre los grupos de pacientes con la mutación y sin esta. Se consideró un nivel de significación de p ≤ 0,05.
RESULTADOS
De los 45 pacientes estudiados, 24 (53,33 %) eran del sexo femenino y 21 (46,66 %) masculinos. De ellos, 30 (66,6 %) tenían color de la piel blanca.
Con respecto a la edad, en la muestra total se encontró una media de 54,5 años con una desviación estándar de 16,2 (rango 18 a 81 años). Llama la atención que el 35,5 % (n = 16) de los pacientes tenían menos de 50 años. Al comparar la edad de los pacientes positivos y negativos para la mutación (57,04 ± 13,01 vs 30,6 ± 11,4 años), se encontró una menor edad en los pacientes negativos para la mutación, lo que resultó ser significativo (p = 0.0000).
En todos los pacientes se excluyó la presencia de causas secundarias y el 100 % fue negativo para el cromosoma Ph o su gen de fusión.
La esplenomegalia fue el signo clínico predominante que se presentó en el 48,8 % del total de pacientes (n = 22) con un porcentaje de expresión mayor en el grupo positivo para la mutación, donde estuvo presente en 21 pacientes (53,84 %).
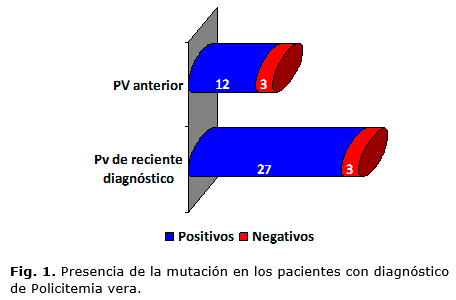
Como se aprecia en la figura 1, la mutación JAK2V617F estuvo presente en el 86,6 % (n = 39) de los pacientes, 12 de ellos con diagnóstico antiguo y en 27 al momento del diagnóstico de la enfermedad.
La eritrocitosis fue confirmada en el 100 % de los pacientes. Cuatro de ellos tenían valores de Hb al diagnóstico inferiores a los recomendados por la OMS para clasificarlos como PV, pero los estudios de volúmenes sanguíneos con cromo 51 confirmaron un índice de masa eritrocitaria superior al 125 %; de ellos 3, habían sido sometidos a flebotomías previas al momento en que se les realizó el estudio radioisotópico. Todos los pacientes negativos para la mutación del JAK tenían menos de 50 años en el momento del estudio molecular.
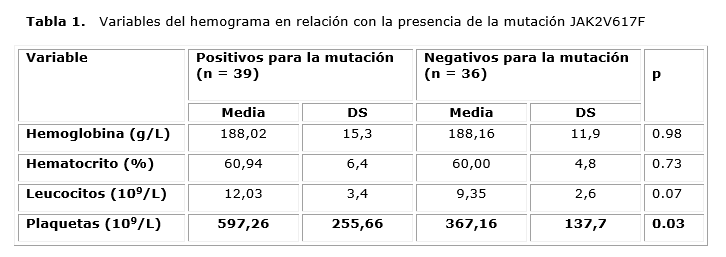
Los resultados de las variables del hemograma (Hb, hematocrito (Hto), conteo de leucocitos y plaquetas) para los pacientes positivos y negativos para la mutación se muestran en la tabla 1. Los resultados de la Hb y el Hto no mostraron diferencias entre ambos grupos y el conteo de leucocitos, aunque fue ligeramente superior en los pacientes positivos para la mutación, no mostró diferencia estadística. Sin embargo, no sucedió así en relación con el conteo de plaquetas, donde los valores de las medias entre ambos grupos sí mostraron diferencias estadísticamente significativas (p = 0,03) y fueron inferiores en los pacientes negativos para la mutación.
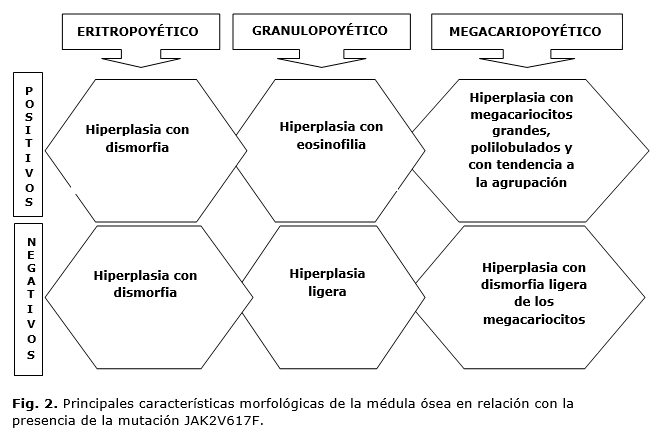
Los estudios de la médula ósea mostraron una médula hipercelular en el 93,3 % de los pacientes (n = 42) y en 44 (97,77 %) se observó panmielosis con algún grado de dismorfia, más evidente en los positivos para la mutación estudiada. Solo en una paciente, cuyo diagnóstico de PV se realizó en la edad pediátrica y que en el momento de este estudio tenía 20 años, se observó eritrocitosis aislada, y en este caso la mutación resultó ser negativa. Las alteraciones más frecuentes para cada línea celular y su relación con la positividad de la mutación JAK2V617F se muestran en la figura 2. Otros datos interesantes son la presencia de eosinofilia moderada en 21 (53,8 %) de los pacientes positivos. En el estudio histológico de la médula ósea, un tercio de los pacientes (n = 15) mostraron incremento de las fibras de reticulina grado I-II.
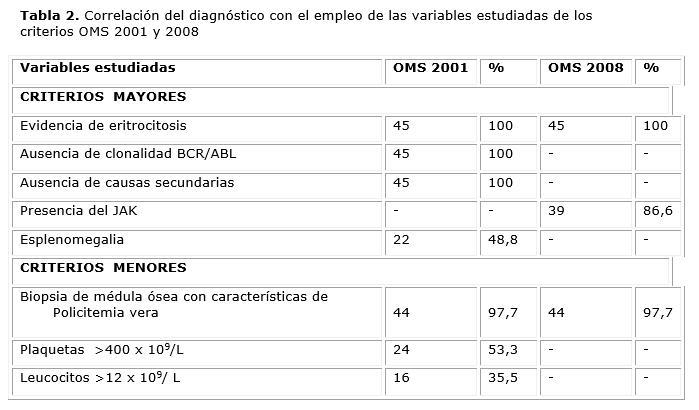
Al realizar la comparación para confirmar el diagnóstico con el empleo de las variables estudiadas para los criterios diagnósticos de la OMS del 2001 y del 2008 (tabla 2) se encontró que se mantuvo el diagnóstico en 39 pacientes.
En los seis pacientes negativos para la mutación, la confirmación no fue posible por ambos criterios debido a la imposibilidad de realizar los otros criterios requeridos y por tanto, no se cumplía con la cantidad de variables requerida para hacer un diagnóstico definitivo según los criterios OMS 2008. Dos de ellos, uno de cada grupo, se concluyeron posteriormente como NMP no clasificadas. Uno, el perteneciente al grupo con diagnóstico anterior, tenía incluso cuantificación de eritropoyetina, realizada en otro país, dentro del rango de referencia. Los otros cuatro pacientes negativos para la mutación estudiada tenían menos de 30 años.
DISCUSIÓN
La PV comúnmente se diagnóstica después de los 40 años, con un pico mayor en los mayores de 70 años. Según algunos autores, la edad media de diagnóstico es alrededor de los 60 años de edad,1,10 lo que no se corresponde exactamente con lo encontrado en el presente estudio. Otros han encontrado una media de edad al inicio de la enfermedad de alrededor de 50 años,11,12 similar a lo encontrado en el presente estudio.
Por otra parte, se plantea que el 5 % de los pacientes con PV son diagnosticados en edades inferiores.1,11,12 En esta casuística, el diagnóstico en menores de 50 años constituyó poco más de la tercera parte de los pacientes estudiados, superior a la mayoría de las series revisadas. Este aspecto pudiera explicarse por el diagnóstico cada vez más temprano de esta enfermedad, debido a la mayor accesibilidad a los servicios de salud en Cuba y a los exámenes médicos periódicos de los trabajadores, ya que muchas veces el diagnóstico se realiza en un estado asintomático y las cifras elevadas de Hb son un hallazgo del chequeo, aspecto que brinda la posibilidad de un diagnóstico precoz.
La PV ha sido descrita con una mayor frecuencia en individuos con ancestros europeos que en aquellos con ancestros africanos,1,11 hecho que se evidencia en el trabajo donde aproximadamente dos tercios de los individuos tenían una apariencia racial blanca, a pesar del gran mestizaje existente en la población cubana.
La esplenomegalia se encuentra entre las principales manifestaciones clínicas de la PV. Según algunos autores, tres de cada cuatro pacientes la presentan al diagnóstico de la enfermedad13, tal es así que desde los criterios diagnósticos establecidos por el Grupo Internacional de Estudios de la PV a inicios de los años 70 del pasado siglo y hasta los de la OMS del 2001, ha sido considerada como un criterio diagnóstico; en el presente estudio constituyó el principal hallazgo clínico encontrado, pero su frecuencia fue inferior a lo descrito en la literatura.4,14,15
La presencia de la mutación JAK2V617F en la PV varía según diferentes estudios realizados, se informa una frecuencia del 65 - 97 % de acuerdo con la sensibilidad del método empleado, el tipo de muestra a utilizar y el uso de leucocitos totales o granulocitos solamente 5,12,16-18. Los resultados encontrados en este estudio, al investigar solo la existencia de la mutación JAK2V617F, están dentro de ese rango, aunque no se corresponden con los porcentajes reportados por la mayor parte de los investigadores. Un aspecto que pudiera influir está en relación con el tamaño de la muestra estudiada, correspondiente a los primeros tiempos de la introducción del estudio molecular en el IHI, ya que la especificidad y sensibilidad de la técnica que se empleó es considerada a la altura de los estándares internacionales.19 En artículos publicados con similar número de casos la positividad de la mutación está entre el 87 y el 90 %.12,20
Se conoce que cerca del 10 % de los pacientes con PV clásica no expresan esta mutación8, y que entre los pacientes negativos para la mutación JAK2V617F se encuentra un grupo que son positivos para otras mutaciones similares, como la del exón 12 del gen JAK descrita por primera vez en el 2007. Actualmente suman más de 10 las mutaciones encontradas en pacientes negativos para el JAK2V617F, que incluyen deleciones, mutaciones puntuales y duplicaciones que afectan mayormente los residuos de aminoácidos altamente conservados (F536–E547), algunas de ellas capaces de producir eritrocitosis en modelos murinos.21,22 La presencia de las mutaciones del exón 12 se han descrito en alrededor del 2 - 5 % de los casos.5,20
La positividad de la mutación JAK2V617F se ha asociado a una mayor edad de diagnóstico de la enfermedad y se señala una media de 59 años12,21, similar a lo encontrado en el estudio. Por otra parte, también se asocia con cifras mayores de Hb y plaquetas, correspondiendo a los homocigóticos las cifras más elevadas.12,17 En los negativos para la mutación se plantea eritrocitosis aislada y son raras la leucocitosis, la trombocitosis y la esplenomegalia.23
En el estudio actual no se encontraron diferencias entre los valores de Hb y Hto en ambos grupos, y aunque se evidenció un ligero incremento en la media del conteo de leucocitos en los pacientes que expresaron la mutación, no se encontró significación estadística. En cambio, coincidiendo con lo descrito en la literatura12, se encontraron conteos de plaquetas superiores en los pacientes positivos para la mutación, lo que desde el punto de vista estadístico si fue relevante. El mayor porcentaje de pacientes con esplenomegalia también correspondió al grupo positivo para la mutación.
En los pacientes negativos para el JAK2V617F se debe buscar la presencia de otras mutaciones del exón 12; estas cursan con una menor edad al diagnóstico, cifras de hemoglobina elevadas, conteos más bajos de leucocitos y plaquetas y ausencia de esplenomegalia.17 Cabe destacar que los pacientes negativos para la mutación clásica correspondieron a pacientes de menor edad en quienes predominó la eritrocitosis y existió una menor alteración en las líneas medulares estudiadas, con ausencia de incremento del retículo.
Aunque en todos los pacientes de esta serie se confirmó la existencia de la eritrocitosis, se coincide con Pearson y Silver 24,25 en que cuando el hematocrito es mayor del 56 % en la mujer y 60 % en el hombre, no es necesario realizar los estudios de volúmenes sanguíneos. Sin embargo, estos pueden ser definitorios en pacientes que tengan valores inferiores a ellos, lo que permitiría establecer un incremento de la masa eritrocitaria mayor del 125 %. Recientemente, algunos investigadores han publicado evidencias de que pudiera existir una policitemia enmascarada o clínicamente latente y que los valores de Hb requeridos por la OMS para el diagnóstico de PV son un indicador exacto de la cantidad de glóbulos rojos en el organismo, sobre todo en pacientes que han sido sometidos a flebotomías o han llevado tratamiento citorreductor con anterioridad26. Sin embargo, la relación entre los niveles de Hb, Hto y la eritrocitosis absoluta medida por los estudios de volúmenes sanguíneos, no está bien definida, debido a que una Hb aumentada no siempre equivale a una eritrocitosis absoluta y una concentración de Hb por debajo del punto de corte requerido para cada género podría corresponder con una índice globular elevado. Por esta razón, los parámetros establecidos por la OMS en cuanto a niveles de Hb como medida de eritrocitosis han sido enjuiciados.27
La biopsia de médula ósea en pacientes con la mutación JAK2V617F se presenta hipercelular con hiperplasia de los tres sistemas y con megacariocitos pleomórficos, gigantes, polilobulados y agrupados1,28; hallazgos comprobados en la biopsia de médula ósea de los pacientes estudiados, a lo que se suma la eosinofilia presente en un porcentaje no desdeñable de los casos estudiados. La presencia en un tercio de los pacientes de incremento de las fibras de reticulita, es un aspecto notorio, ya que esto se asocia con la progresión a mielofibrosis y también es más frecuente en los pacientes con presencia de clonalidad.17,25
En los pacientes con mutaciones del exón 12 predomina, sobre todo, la hiperplasia del sistema eritropoyético, el granulopoyético puede estar moderadamente hiperplásico y los megacariocitos pueden estar aumentados, pero sin la agrupación característica encontrada en los pacientes positivos28, hecho que en este estudio se presentó con más frecuencia en los pacientes en los que no se encontró la mutación estudiada.
Estos resultados demuestran que es posible realizar el diagnóstico de la PV con el empleo de los criterios de la OMS del 2001, pero el estudio molecular, facilita la precisión diagnóstica y contribuye a reducir el margen de error diagnóstico en la PV, sobre todo en fases tempranas de la enfermedad; sin embargo, coincidimos con otros autores en que se hace necesaria la revisión de los criterios diagnósticos de la OMS 2008 25,26, pues el diagnóstico no debe basarse tan sólidamente en la presencia de la mutación del JAK2 o la ausencia de esta, ya que la mutación está presente en varias NMP e incluso en otras malignidades. Por otra parte, los niveles de eritropoyetina pueden estar normales hasta en el 20 % de los pacientes y los cultivos eritroides son muy poco utilizados en el mundo como parte de la rutina diagnóstica de la PV 29.
Por nuestra parte consideramos que la biopsia de médula ósea debe constituir uno de los criterios mayores ya que puede contribuir a diferenciar la PV de las otras neoplasias mieloproliferativas e incluso, de cambios reactivos. Además, se deben incluir entre los criterios menores algunos elementos clínicos como la esplenomegalia, demostrable tanto por la palpación como por ultrasonografía. Una revisión de los criterios actuales con bases sólidas y evaluadas objetivamente podría garantizar un diagnóstico certero en cualquier lugar del mundo.
Agradecimientos
A los profesionales y técnicos de los laboratorios de Biología Molecular, Anatomía Patológica y Citomorfología del Instituto de Hematología e Inmunología, por su cooperación en la realización e informe de los estudios moleculares, histológicos y los hemogramas, respectivamente
REFERENCIAS BIBLIOGRÁFICAS
1. Kremyanskaya M, Najfeld V, Mascarenhas J, Hoffman R. The polycythemias. In Hoffman R, Benz EJ Jr., Silberstein LE, Heslop HE, Weitz JI, eds. Hematology basic principles and practice. 6th ed. Philadelphia: Elsevier Saunders; 2013.p. 988-1033.
2. Fernández-Jiménez MC, Murga MJ, Remacha AF. Clasificación y diagnóstico de las eritrocitosis. Haematologica/edición española. 2010;95(Extra 1):423-8.
3. Chen E, Beer P, Godfrey A, Ortmann C, Lee J, Costa-Pereira A, et al. Distinct clinical phenotypes associated with JAK2V617F reflect diferential STAT1 signaling. Cancer Cell. 2010 Nov;18(5):524-35. doi: 10.1016/jccr.2010.10.013.
4. Roda P, Ferrari A, Tang X, Erlich P, Eisenhower C, Patel MD et al. Determination of accuracy of polycythemia vera diagnoses and use of the JAK2V617F test in the diagnostic scheme. Ann Hematol. 2014 Sep;93(9):1467-72. doi: 10.1007/s00277-014-2068-2.
5. Muxí PJ, Oliver AC. JAK-2 Positive Myeloproliferative Neoplasms. Curr Treat Options Oncol. 2014 Jun;15(2):147-56. doi: 10.1007/s11864-014-0279-3.
6. Ugo V, Tondeur S, Menot ML, Bonnin N, Le Gac G, Tonetti C, et al. Interlaboratory development and validation of a HRM method applied to the detection of JAK2 exon 12 mutations in polycythemia vera patients. PLoS One. 2010; 5(1): e8893. doi: 10.1371/journal.pone.0008893.
7. Vardinman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009 Jul; 114(5): 937-51. doi: 10.1182/blood-2009-03-209262.
8. Fernández Delgado N, Fundora Sarraff T, Macias Pérez I. Policitemia vera. Experiencias en el diagnóstico y tratamiento en el Instituto de Hematología e Inmunología. Rev Cubana Hematol Inmunol Hemoter. 2011:27(1)77-90.
9. Means R. JAK2 V617F and the evolving paradigm of polycythemia vera. Korean J Hematol. 2010 Jun; 45(2): 90–4. doi: 10.5045/kjh.2010.45.2.90.
10. Caires dos Santos L, Correa da Costa J. Pereira N, Cerutti J, Regis da Silva M, et al. Cytogenetic, JAK2 and MPL mutations in polycythemia vera, primary myelofibrosis and essential thrombocythemia Rev Bras Hemato. Hemoter. 2011; 33(6):417-24. DOI: 10.5581/1516-8484.20110116.
11. Ruíz-Argüelles G, Ruíz-Delgado G, Ruíz-Reyes G. Síndromes mieloproliferativos crónicos. En: Palomo I, Pereira J, Palma J, eds. Hematología: Fisiopatología y Diagnóstico. Talca:Universidad de TALCA; 2005.p.320-32.
12. Scott LM. The JAK2 exon 12 mutations: A comprehensive review. Am J Hematol. 2011 Aug; 86(8):668-76. doi: 10.1002/ajh.22063.
13. González García C, Funes Vera C, Blanquer Blanquer M, Moraleda Jiménez JM. Síndromes mieloproliferativos Medicine. 2012;11(21):1289-97.
14. Tefferi A, Rumi E, Finazzi G, Gisslinger H, Vannucchi AM, Rodeghiero F et al. Chronic Myeloproliferative Neoplasias. Survival and prognosis among 1545 patient with contemporary polycythemia vera: an international study. Leukemia. 2013 sep; 27(9):1874-81. doi: 10.1038/leu.2013.163.
15. Martín Pueyo G, Bello J. Policitemia vera. FMC. 2011;18(1):8-12
16. Amor A, Díaz C, Garrote H, Suárez Y, Fernández N, et al. Introducción del estudio molecular de la mutación JAK2V617F en neoplasias mieloproliferativas clásicas BCR-ABL negativas. Rev Cubana Hematol Inmunol Hemoter. 2013;29(4):398-406.
17. Sonbol M, Firwana B, Zarzour A, Morad M, Rana V. et al. Comprehensive Review of JAK Inhibitors in Myeloproliferative Neoplasms. Ther Adv Hem. 2013 Feb; 4(1):15-35. doi: 10.1177/2040620712461047.
18. Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010 Sep; 363(12):1117–1127. doi: 10.1056/NEJMoa1002028.
19. Veneri D, Capuzzo E, de Matteis G, Franchini M, Baritono E, Benati M, et al. Comparison of JAK2V617F mutation assessment employing different molecular diagnostic techniques. Blood Transfus. 2009 Jul; 7(3):204-9.doi: 10.2450/2009.0070-08.
20. Siemiatkowska A, Bieniaszewska M, Hellmann A, Limon J. JAK2 and MPL gene mutations in V617F-negative myeloproliferative neoplasms. Leuk Res. 2010 Mar; 34(3):387–9. doi: 10.1016/j.leukres.2009.06.017
21. Tefferi A. Novel mutations and their functional and clinical relevance in myeloproliferative neoplasms: JAK2, MPL, TET2, ASXL1, CBL, IDH and IKZFz. Leukemia. 2010 Jun; 24(6): 1128–38. doi:10.1038/leu.2010.69.
22. Laughlin T, Moliterno A, Stein B, Rothberg P. Detection of Exon 12 Mutations in the JAK2 Gene Enhanced Analytical Sensitivity Using Clamped PCR and Nucleotide Sequencing. J Mol Diagn. 2010 May; 12(3): 278–82. doi:10.2353/jmoldx.2010.090177.
23. Schnittger S, Bacher U, Haferlach C, Geer T, Müller P, et al. Detection of JAK2 exon 12 mutations in 15 patients with JAK2V617F negative polycythemia vera. Haematologica 2009 Mar; 94(3):414-8. doi:10.3324/haematol.13223.
24. Pearson TC. Apparent polycythaemia. Blood Rev.1991; 5(4):205-13.
25. Silver RT, Chow W, Orazi A, Arles SP, Goldsmith SJ. Evaluation of WHO criteria for diagnosis of polycythemia vera: a prospective analysis. Blood. 2013 Sept; 122(11):1881-6.
26. Barbui T, Thiele J, Gisslinger H, Finazzi G, Carobbio A, Rumi E, et al. Masked polycythemia Vera (mPV): Results of an international study. Am J Hematol.2014 Jan; 89(1): 52–4. doi:10.1002/ajh.23585.
27. Johansson PL, Safai-Kutti S, Kutti J. An elevated venous haemoglobin concentration cannot be used as a surrogate marker for absolute erythrocytosis: A study of patients with polycythaemia vera and apparent polycythaemia. Br J Haematol; 2005 Jun; 129(5):701–5.
28. Lakey MA, Pardanani A, Hoyer JD, et al. Bone marrow morphologic features in polycythemia vera with JAK2 exon 12 mutations. Am J Clin Pathol. 2010 Jun;133(6):942-8. doi: 10.1309/AJCP3Z2AKUWRGTNM.
29. Kvasnicka HM. WHO Classification of Myeloproliferative Neoplasms
(MPN): A Critical Update. Curr Hematol Malig Rep. 2013; 8:333– 41 DOI: 10.1007/s11899-013-0186-x.
Recibido: Octubre
29, 2014
Aceptado:
Enero 07, 2015
Dra. Norma D. Fernández Delgado . Instituto de Hematología e Inmunología. Apartado 8070, La Habana, CP 10800, CUBA. Tel (537) 643 8695, 8268. Email: rchematologia@infomed.sld.cu
{kind=link}
{kind=link}
{kind=link}