ARTÍCULO DE REVISIÓN
Hemoglobinuria paroxística nocturna: de Strübing al Eculizumab
Paroxysmal nocturnal hemoglobinuria: from Strübing to Eculizumab
Dra. Ivis Macía Pérez, Dra. Tania García Peralta, Ing. Teresa Fundora Sarraff, Dra. Norma Fernández Delgado
Instituto de Hematología e Inmunología
RESUMEN
La hemoglobinuria paroxística nocturna (HPN) es un trastorno clonal severo y raro no maligno y adquirido de la célula madre hematopoyética. Es el único trastorno hemolítico adquirido causado por una anomalía de la membrana eritrocitaria como resultado de una mutación somática clonal de un gen, el fosfatidilinositol glucano clase A (PIG-A) situado en el brazo corto del cromosoma X. Se han identificado una serie de proteínas reguladoras del complemento, entre las que se destacan: el factor acelerador de la degradación (CD55) y el factor inhibidor de la lisis reactiva de la membrana (CD 59) deficientes en esta enfermedad. La HPN se clasifica en clásica, asociada a otro trastorno medular y en subclínica. Su diagnóstico se apoya en estudios hematológicos, bioquímicos, pruebas serológicas especiales, estudios eritroferrocinéticos e imagenológicos. La electroforesis de proteínas de membrana de alta resolución y la citometría de flujo multiparamétrica constituyen técnicas de elección para el diagnóstico. El tratamiento de la anemia, de los episodios trombóticos y de las infecciones constituyen los pilares terapéuticos básicos. Dentro de los agentes farmacológicos más utilizados se destacan: los esteroides. los andrógenos, la eritropoyetina recombinante humana y el factor estimulador de colonias granulocíticas. Recientemente, el anticuerpo monoclonal eculizumab ha aumentado la expectativa de vida de estos pacientes con una mejoría de su calidad de vida.
Palabras clave: hemoglobinuria paroxística nocturna, fosfatidilinositol glucano clase A, citometría de flujo multiparamétrica, eculizumab.
ABSTRACT
Paroxysmal nocturnal hemoglobinuria (PNH) is a non malignant and acquired clonal disease of the hematopoietic stem cell. It is a severe and rare disease. It is the only acquired hemolytic disturbance that is caused for an erythrocyte membrane anomaly. It is a result of a somatic clonal mutation of one gene that is located in the short arm of X chromosome called phosphatidyl inositol glycan class A (PIG-A). Regulated complement proteins are identified: the decay accelerated factor (CD55) and the membrane inhibitor or reactive lysis (CD 59); the abnormal blood cells of PNH have deficiency of these two proteins. PNH is classified in: classic PNH, PNH associated with another bone marrow disturbance and PNH sub clinic. Diagnosis is obtained by hematological, biochemical, kinetics and imagenologics studies and serologic special tests. High resolution membrane protein electrophoresis and flow cytometry are the elective tests. Treatments for anemia, thrombotic episodes and infections are important in the management of these patients. Steroids, androgens, human recombinant erythropoietin and granulocytic colony stimulating factor (CSF-G) are the more used pharmacology agents. Recently, the monoclonal antibody eculizumab has increased the life expectation in these patients with a better quality of life.
Key words: paroxysmal nocturnal hemoglobinuria, phosphatidylinositol glycan class A, multiparameter flow cytometry, eculizumab.
INTRODUCCION
La hemoglobinuria paroxística nocturna (HPN), es una anemia hemolítica crónica adquirida causada por un trastorno clonal de la célula madre hematopoyética.1 Es una enfermedad rara y con gran variabilidad clínica, con una incidencia de 0,05-0,13 casos por cada cien mil habitantes por año.2 La mortalidad es del 50 % con una mediana de supervivencia de 22 años. Más del 60 % de las muertes son causadas por trombosis y hemorragias.1,2
ANTECEDENTES HISTÓRICOS
Como se pensaba que la entidad se había descubierto por Marchiafava y Nazari en 1911 y se describió más detalladamente por Micheli en 1931, se le denomina también como Síndrome de Marchiafava-Micheli.2-4 En realidad, la enfermedad se conocía desde el siglo XIX, pues Strûbing hizo una excelente descripción de ella en 1882 4 y Hegglin, Maier y Ham también realizaron aportes de interés.5 Ya en 1945 se habían registrado alrededor de 46 casos, en 1953 se conocían 162 enfermos y diez años más tarde Dacie comunicó 48 pacientes más con esta entidad.4
En 1920, Adolfo Ferrata expuso que Effore Marchiafava fue el que denominó la enfermedad como hemoglobinuria paroxística nocturna con hemosideruria perpetua. 2,3 Desde esa época hasta la actualidad se ha mantenido el criterio de que en estos pacientes ocurre una hemoglobinuria macroscópica que puede tener períodos de exacerbaciones.2 El término nocturna es una reliquia histórica, por haberse descrito los primeros casos de hemoglubinuria tomando en consideración la aparición de orinas oscuras en los enfermos por la noche o al despertar.3,4 En realidad, esta alteración está relacionada con una crisis hemolítica importante que ocurre en los enfermos durante el sueño.3,4
ETIOPATOGENIA
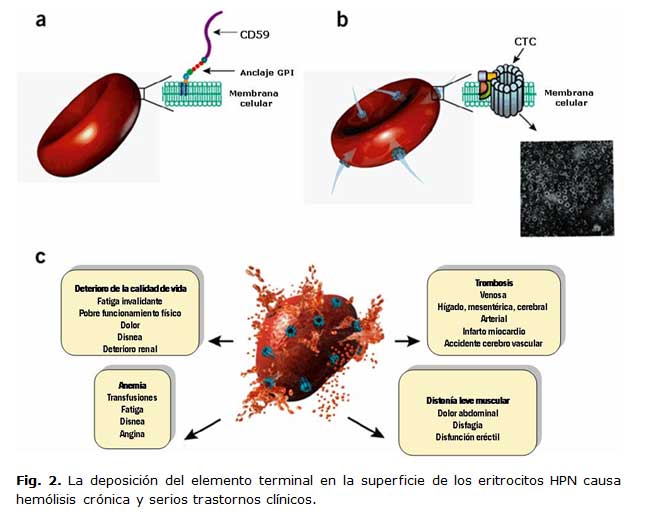
La HPN es un trastorno hemolítico adquirido donde la lesión primaria es producida por una mutación somática y clonal de la célula madre hematopoyética en la biosíntesis de glucosilfosfatidilinositol (GPI). La carencia total o parcial de la expresión de las proteínas de la membrana ancladas a través del GPI provocan una sensibilidad anormal al complemento.1,3 (Figura 1)
En la biosíntesis del anclaje al GPI están involucradas al menos 10 reacciones y más de 20 genes diferentes. Dos de las proteínas, el factor acelerador de la degradación (DAF, CD 55) y el factor inhibidor de la lisis reactiva de membrana (ILRM, CD 59) que inhiben la activación y función citolítica del complemento, respectivamente, son postuladas como las de mayor importancia en la fisiopatología de la enfermedad.1-3,6
Los eritrocitos de la HPN se han clasificado tradicionalmente en subpoblaciones, según su susceptibilidad a la lisis mediada por complemento.7-9
Los primeros defectos observados en la superficie de las células sanguíneas maduras en esta enfermedad fueron la disminución de la acetilcolinesterasa en los hematíes y de la fosfatasa alcalina en los leucocitos.2 En la actualidad, ya son más de 20 las proteínas de membrana cuya expresión se ha encontrado disminuida o ausente. 3, 5-13 De ellas sólo 6 tienen trascendencia clínica (Tabla 1) y la expresión de la enfermedad depende del tipo de proteína de membrana que falta y del grado de la alteración de su función. 3, 5, 6, 9-16
Tabla 1. Prote�nas deficientes en las c�lulas de HPN
|
P D D |
Factor acelerador de la degradaci�n (DAF, CD 55) |
Expresi�n en: Eritrocitos, neutr�filos y monocitos |
|
Funci�n: Control de la activaci�n
del complemento en la superficie celular. |
||
|
Consecuencia del defecto: Mayor fijaci�n de complemento por las c�lulas� y la hemolisis intravascular |
||
|
Factor inhibidor de la lisis reactiva de membrana (ILRM, CD 59) |
Expresi�n en: Todas las l�neas celulares hematopoy�ticas. |
|
|
Funci�n: Bloqueo del ensamblaje
del complejo de ataque a la membrana (C5b9) |
||
|
Consecuencia del defecto: Hemolisis intravascular |
||
|
Factor de restricci�n
hom�loga |
Expresi�n en: Membrana eritrocitaria |
|
|
Funci�n: Se une al C8 y demuestra la restricci�n de la especie. |
||
|
Consecuencia del defecto: Hem�lisis por lisis mediada por complemento |
||
|
R |
�Receptor Fcg III a (CD 16) |
Expresi�n en:� Neutr�filos |
|
Funci�n: Prepara los neutr�filos para la fagocitosis |
||
|
Consecuencia del defecto: Susceptibilidad a las infecciones |
||
|
Receptor activador del plasminogeno tipo� uroquinasa (RAP u) |
Expresi�n en : Plaquetas |
|
|
Funci�n: Activa el plasmin�geno a plasmina e inicia la fibrinolisis |
||
|
Consecuencia del defecto: Favorece fen�menos tromb�ticos |
||
|
�Campath-1� (CDw52) |
Expresi�n en : Linfocitos, monocitos y neutr�filos |
|
|
Funci�n: Activaci�n linfocitaria T por v�a CD2 |
||
|
Consecuencia del defecto: Favorece activaci�n del complemento y por tanto la lisis de la c�lula. |
En la ultima década diferentes investigadores han demostrado la expresión de la proteína prion (PrPc) en células hematopoyéticas humanas y se ha sugerido que podría estar involucrada en el funcionamiento del sistema hematopoyético. Esta proteína, bien conocida por su participación en la patogenia de un grupo de enfermedades cerebrales transmisibles, es una glicoproteína que está anclada a la superficie externa de la membrana por la unión al GPI y su expresión se ha relacionado con la activación y desarrollo de las células hematopoyéticas. La expresión de la PrPc se ha encontrado disminuida en la superficie de las células CD34 positivas y en los leucocitos de sangre periférica de pacientes con HPN y se ha comprobado que las plaquetas activadas de estos pacientes expresan la PrPc .3, 5, 6, 9-16
CUADRO CLÍNICO
Es una entidad poco frecuente que afecta ambos sexos y aparece entre los 16 y 75 años, con una edad promedio de 42 años, aunque se ha descrito también en niños y en ancianos. Su comienzo es insidioso y su evolución es prolongada y variable.1,3,5
En la figura 2 se muestra como el depósito del complemento terminal en la superficie de los eritrocitos HPN ocasionan la hemólisis crónica y trastornos clínicos severos.
La sintomatología de la HPN está dada por astenia, dolor subesternal, abdominal o lumbar, somnolencia, malestar general, fiebre y cefaleas intensas, que pueden persistir por varios días. El dolor abdominal puede ser a tipo cólico y prolongarse de uno a dos días.1-3,6
Los signos clínicos no son muy manifiestos, aunque puede constatarse palidez cutáneo-mucosa, íctero y coloración bronceada de la piel en relación con las múltiples transfusiones que reciben algunos de ellos, en dependencia del patrón clínico. Puede existir esplenomegalia moderada y en ocasiones, hepatomegalia de ligera a moderada, aunque en muchos enfermos el examen físico suele ser normal. 2,5
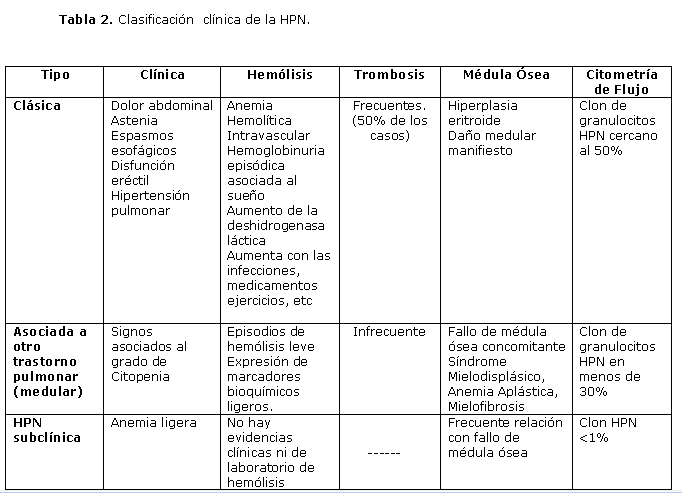
La enfermedad se clasifica de acuerdo a la clínica en tres grupos: 2,3
HPN clásica: 1-3, 6 HPN asociada a otro trastorno medular10,12,17-21y HPN Subclínica2,3 (Tabla 2).
En los pacientes
con HPN son frecuentes las infecciones por defectos cualitativos, cuantitativos
o ambos de los leucocitos y linfocitos HPN, por una elevada sensibilidad a
la lisis mediada por el complemento y disminución de la expresión
de las proteínas de membrana, como la Fc gamma RIIIa, CD16 y CD 55
que trae como resultado una deficiencia en la quimiotaxis y la fagocitosis.2,22,23
Además pueden aparecer complicaciones hemorrágicas graves que
ocupan el segundo lugar en las muertes de estos casos.2, 22, 23
Las alteraciones renales que aparecen son: la hematuria, la proteinuria, la hipertensión y el deterioro de la concentración de la orina que pueden llegar hasta la insuficiencia renal aguda y crónica en algunos casos.2, 22, 23
DIAGNÓSTICO DE LABORATORIO
Por el cuadro clínico abigarrado que presenta y la variabilidad en la positividad de las pruebas que se utilizan para su identificación, esta enfermedad resulta en muchas ocasiones muy difícil de diagnosticar.
Los estudios hematológicos muestran una anemia variable que en algunos pacientes puede ser severa, de hasta 50 g/L con un patrón de hemólisis, leucopenia y trombocitopenia con recuento de reticulocitos elevado; exceptuando al patrón clínico que se asocia con aplasia medular (AM). Los sangramientos frecuentes y la hemosideruria pueden llevar a ferropenia. Los estudios de la coagulación y dentro de ellos los de hipercoagulabilidad son imprescindibles, porque estos pacientes se incrementa el riesgo trombótico.1,3,6,24
La citometría de flujo multiparamétrica constituye la técnica de elección para el diagnóstico de la HPN por su gran sensibilidad y especificidad.25-28 En el 2009, un grupo de expertos españoles llegaron a un consenso para el empleo de la citometría de flujo en el estudio de los pacientes sospechosos de HPN. La misma debe realizarse en pacientes con anemia hemolítica no inmune y hemoglobinuria y en pacientes con trombosis en sitios infrecuentes.1 En la HPN, el estudio de la expresión de moléculas asociadas a GPI debe realizarse en al menos dos líneas celulares empleando 2 anticuerpos monoclonales diferentes.
Cuando se marcan solamente los eritrocitos pueden existir falsos positivos, sobre todo si recientemente ha ocurrido un episodio hemolítico o una transfusión. Los monocitos y granulocitos tienen una vida media más corta y no se afectan por las transfusiones, por lo que el porcentaje de células HPN en estas líneas refleja mejor el tamaño del clon.
Los marcajes más utilizados son el CD16 (Fc gamma RIIIB), CD 24, CD 66, CD 55 y CD 59 en neutrófilos; CD 14, CD 55 y CD 59 en monocitos y CD 59 y CD 55 en hematíes. De menor utilidad se encuentran el CDw52 en linfocitos y monocitos, el CD 87 en neutrófilos, linfocitos T activados y monocitos o CD 48 en linfocitos y monocitos.
Se recomienda el marcaje simultáneo de los hematíes con CD 55 y CD 59 y puede añadirse el CD 235 (Glicoforina A) para seleccionar la población eritroide. El CD 59 es el que mejor permite identificar las subpoblaciones de hematíes de HPN.2, 25-28
Esta técnica constituye un examen indispensable para el diagnóstico de esta enfermedad, la evaluación inicial del riesgo hemolítico y trombótico, el cálculo del tamaño clonal al debut y en su evolución posterior al tratamiento con eculizumab. La citometría de flujo también esta indicada en los enfermos con HPN clásica sin tratamiento y con HPN subclínica con frecuencia anual.2, 25-28
La Electroforesis de proteínas de membrana de alta resolución estudio basado en la identificación de los defectos proteicos de la membrana celular mediante el uso de anticuerpos monoclonales contra los antígenos CD55 y CD59, confieren el mejor panel para definir las poblaciones celulares del Tipo I (Normal), del Tipo II (Deficiencia parcial de GPI) y del Tipo III (Deficiencia total de GPI). 27
Este método es sensible y específico, y puede brindar información en cuanto a la proporción de poblaciones celulares anormales. En los últimos años la identificación de las proteínas de membrana afectadas en los pacientes con HPN, mediante esta técnica ofrece la posibilidad de identificar exactamente la proteína deficiente y su cuantía. Estos estudios son altamente costosos lo que limita su empleo rutinario.27
Hasta el momento, la demostración de las proteínas de membrana carentes, ligadas al GPI en glóbulos rojos y granulocitos mediante el uso de anticuerpos monoclonales (CD55 y CD59) y la citometría de flujo, es la mejor forma de diagnosticar la HPN.
OTROS EXÁMENES COMPLEMENTARIOS ÚTILES
El diagnóstico
de la HPN se apoya también en estudios hematológicos que incluyen
el estudio de médula ósea, estudios enzimáticos y bioquímicos
(LDH bilirrubina indirecta, haptoglobina) y pruebas serológicas especiales
(test de Ham Crosby y Sucrosa). 1,3,6, 8, 24
En nuestro medio los estudios eritroferrocinéticos también han sido de gran ayuda para el diagnóstico de la HPN, sobre todo cuando esta se presenta como una anemia de etiología no precisada.8, 23
Los estudios de imágenes (ecografía Doppler abdominal, 2 ecocardiografía Doppler y angio-TAC pulmonar, TAC y resonancia magnética nuclear) pudieran ser útiles en la detección precoz de eventos tromboticos a diferentes niveles. 2
TRATAMIENTO
Desde el punto de vista práctico, el tratamiento de la HPN se divide en tres aspectos fundamentales: 1,3, 6,12, 20,21
· Tratamiento de la anemia.
· Prevención y tratamiento de la trombosis.
· Modificadores de la hematopoyesis.
Tratamiento de la anemia
Los glucorticoides constituyen la droga de elección para disminuir la hemólisis y prevenir la anemia, su mecanismo de acción puede inhibir la activación del complemento por vía alternativa o estabilizar la membrana eritrocitaria. Las dosis son relativamente altas,1-2 mg/kg/día. 1, 3, 6, 12, 20, 21
Las transfusiones de glóbulos rojos también son útiles para controlar la anemia y mejorar la hemólisis, porque provocan un freno de la eritropoyesis y con ello de la producción de eritrocitos sensibles al complemento. Durante años se planteó que las transfusiones debían ser con glóbulos lavados, pero no hay evidencias de que esta modalidad evite la acción del complemento aportado por el plasma. La recomendación mas aceptada en la actualidad es el uso de glóbulos rojos desleucotizados o filtrados para evitar que posibles antígenos-anticuerpos estimulen el complemento y potencien la hemólisis.1, 3, 6,12, 20, 21
Prevención y tratamiento de la trombosis
La efectividad de la anticoagulación preventiva no ha sido probada en pacientes con HPN y frecuentemente puede resultar peligrosa debido a la trombocitopenia que acompaña a una buena parte de los pacientes. Su uso se justifica solo en pacientes con niveles elevados de dímero D, durante la gestación y en el preoperatorio.29
La trombosis aguda
es una urgencia que debe ser tratada con agentes trombolíticos como la
estreptoquinasa, la uroquinasa y el activador tisular del plasminógeno,
siempre que no existan contraindicaciones para su uso.6,12, 20,
21
Durante la trombosis aguda el paciente debe recibir heparina de bajo peso molecular a la dosis habitual, seguido por el monitoreo cuidadoso de los parámetros de anticoagulación.6,12, 20, 21
Modificadores de la hematopoyesis
Los andrógenos pueden estimular la hematopoyesis en algunos pacientes, estos son más efectivos en los casos de hipoplasia medular. Los más utilizados son: danazol, fluoximesterona y la oximetalona. Recientemente, se han utilizado citoquinas recombinantes como la eritropoyetina y el factor estimulador de colonias granulocitos (FEC-G). 6,12, 20, 21
Transplante de médula ósea
El transplante de médula ósea es aun en nuestros días el único tratamiento curativo para la HPN, sin embargo está asociado con una elevada morbimortalidad.29-31 Los resultados de estudios no randomizados han sugerido que se pueden lograr remisiones a largo plazo con transplante de células madre hematopoyéticas (TCMH) alogénico. En los pacientes con complicaciones de la HPN con riesgo para su vida el TCMH no mieloablativo o de intensidad reducida podría ser más apropiado, que con el régimen mieloablativo, el que se ha asociado con una mayor mortalidad posterior al transplante. La decisión de hacer el TCMH debe tener en cuenta el pronóstico de la enfermedad incorporando los factores pronósticos adversos conocidos, como la historia previa de trombosis, y la evolución hacia la pancitopenia o el síndrome HPN- Aplasia; frente al riesgo que conllevan las complicaciones del transplante. 32
Eculizumab 29, 30, 32, 33
El eculizumab, anticuerpo IgG2/4k monoclonal humanizado, producido mediante tecnología de ADN recombinante que actúa inhibiendo selectivamente la proteína C5 del complemento humano e impidiendo la formación del complejo terminal, responsable de la formación de canales que causan la lisis del eritrocito, ha aumentado la expectativa de vida de los pacientes con HPN. 2,29, 30, 34-37
Este es bien tolerado y logra la eliminación de la hemólisis, por lo que mejora los niveles de hemoglobina y disminuye los requerimientos transfusionales. Además mejora la sintomatología clínica en relación con la distonía del músculo liso. El eculizumab disminuye, además, la incidencia de trombosis, mejora la función renal y disminuye la hipertensión pulmonar. 2,29, 30, 34-43
En un 10 % de los pacientes que han recibido eculizumab se observó una respuesta subóptima debido a la persistencia de la hemólisis de tipo extravascular, ya que este medicamento no actúa sobre la fracción C3 del complemento que se deposita sobre los eritrocitos. 2
Por su mecanismo de acción, el eculizumab aumenta la sensibilidad para bacterias encapsuladas, fundamentalmente la Neisseria, por lo que es necesario vacunar contra la meningitis a todos los pacientes dos semanas antes de la administración del medicamento. La reactivación es necesaria para prevenir esta infección. 2,29, 30, 34-37,44
La dosis del eculizumab es de 600 mg una vez por semana por vía intravenosa durante 4 semanas. La quinta semana la dosis es de 900 mg y a partir de ese tiempo se administran 900 mg cada 14 días de forma ininterrumpida. 2, 38-43
Este novedoso tratamiento produce estabilización de los niveles de hemoglobina y mejoría en la calidad de vida del paciente, pero no cura la HPN. 32
A pesar de sus ventajas, el eculizumab es un medicamento con un costo elevado, que debe utilizarse ininterrumpidamente una vez que se inicia su uso y que no elimina el clon HPN, por lo que se sugiere reservarlo para pacientes muy sintomáticos, con un gran porcentaje de células HPN o con trombosis anteriores. 2
REGISTRO DE HEMOGLOBINURIA PAROXÍSTICA NOCTURNA
El Registro Internacional de HPN es parte del plan de gestión de riesgos exigido por la Federación de Drogas y Alimentos y la Agencia Europea del Medicamento tras la aprobación del eculizumab. 2 Su objetivo es evaluar la eficacia y la seguridad a largo plazo de este fármaco con especial atención en la incidencia de infecciones graves, hemólisis tras la interrupción del tratamiento, reacciones en la infusión o en la administración, aparición de neoplasias secundarias y complicaciones durante la gestación y en neonatos. Sus datos ayudarán a definir la progresión de la enfermedad, la respuesta clínica y la morbimortalidad en pacientes tratados y no tratados con eculizumab. El registro recoge datos analíticos y del tamaño del clon de HPN en hematíes y granulocitos medidos por citometría de flujo.
Hasta nuestros días, el eculizumab es el único medicamento aprobado por la FDA (Food and Drug Administration) para el tratamiento de la HPN. 2 Su introducción ha logrado que aun sin curar la enfermedad los pacientes con HPN no sufran crisis de hemoglubinuria, sean afectados por una menor cantidad de síntomas molestos, no dependan de transfusiones y tengan un menor riesgo de trombosis venosa. De acuerdo con expertos en el estudio y tratamiento de la HPN, los esfuerzos en el área terapéutica en los próximos años estarán encaminados a encontrar vías para controlar la hemólisis extravascular que se presenta en otro grupo de pacientes con HPN. Más allá permanece el reto de encontrar algún tratamiento que además de controlar, cure la HPN. 2
Las opciones terapéuticas a largo plazo incluyen los agentes inmunosupresores como la Globulina Antitimocítica o antilinfocitica más ciclosporina, especialmente en pacientes con citopenias severas o moderadas, mientras que en la era del eculizumab el TCPH podría ser reservado solo para pacientes que desarrollen un Síndrome Mielodisplásico. 45
HPN Y EMBARAZO
El embarazo en mujeres con HPN es raro, con muy pocos reportes de las tasas de mortalidad materna y fetal 46. En realidad a las mujeres jóvenes con HPN generalmente se les aconsejaba que no se embarazaran, ya que en este período se podría empeorar la anemia y más aun debido al riesgo de trombosis.30 Las necesidades de suplementos de hierro y folatos en estas embarazadas son aun mayores debido a la hemólisis intravascular que presentan.29
Una recomendación en el tratamiento de la embarazada con HPN es el uso de la heparina de bajo peso molecular durante todo el embarazo y el puerperio, las transfusiones de glóbulos rojos de ser necesarias y la vigilancia de la aparición de signos y síntomas de trombosis venosa, que incluyen la etapa post parto.30
En un estudio multicéntrico que incluyó mujeres con HPN embarazadas que no habían sido tratadas con eculizumab las complicaciones encontradas fueron las citopenias, con requerimientos transfusionales en el 95 % de los casos, y la trombosis post parto que fue fatal en algunos casos. En el grupo estudiado se presentó partos pretérmino en el 29%, y el peso al nacer fue menor a los 3kg en el 53% de los casos. La mortalidad fetal estuvo alrededor del 4%. 46
El uso del eculizumab ha mejorado el curso clínico, evolución y pronóstico del embarazo en pacientes con HPN y los estudios publicados al respecto indican la seguridad y efectividad en esta etapa. 29, 30, 34
EVOLUCIÓN
El proceso hemolítico crónico intravascular puede llevar a estos pacientes a una deficiencia de hierro, por ello su seguimiento es importante, ya que permitirá instaurar un tratamiento con sales ferrosas para no afectar la eritropoyesis compensatoria. Algunos pacientes evolucionan a un síndrome de AM con altos requerimientos transfusionales que puede ocasionar hemocromatosis secundaria. Se ha descrito un 15 % de recuperación espontánea sin ningún tipo de secuelas después de haber padecido la enfermedad.42-45 La transformación leucémica del clon HPN es muy rara, se señala en un 4 % de los casos. 42-45
Se han comunicado casos de asociación de SMD / HPN aunque son muy discutidos, también se señala la coexistencia con síndromes linfoproliferativos y es extremadamente rara la asociación con otros síndromes mieloproliferativos.1, 3, 6, 12, 20, 21
Con el empleo de la citometría de flujo se ha descrito la detección de pequeños clones de HPN en pacientes con AM o SMD, incluso en ausencia de hemólisis. 2, 28, 46 - 48
SEGUIMIENTO
Debido a las complicaciones hemorrágicas, infecciosas y trombóticas asociadas, estos pacientes deben contar con un seguimiento hematológico estricto y periódico que incluya exámenes complementarios trimestrales (hemograma, conteo de reticulocitos, función renal y hepática, LDH, haptoglobina plasmática, hemosiderina en orina, perfil férrico y prueba de coombs.), y semestrales o anuales (citometría de flujo, estudios de médula ósea y pruebas imagenológicas ( Doppler abdominal y ecocardiografía). 5,7,49 y 50
PRONÓSTICO
La HPN es una enfermedad de curso crónico con una sobrevida media de 10 a 15 años. El 25 % de los enfermos sobreviven 25 años después de realizado el diagnóstico 1, 3,12, 20, 21 lo que podría variar con los efectos del eculizumab donde ya se ha demostrado mejoría evidente en manifestaciones clínicas y calidad de vida.
REFERENCIAS BIBLIOGRAFICAS
1. Morado M, Subirá D, López M. Hemoglobinuria paroxística nocturna: nuevos tratamientos y recomendaciones generales para su diagnóstico. Med Clin 2010; 134(8):369-74.
2. Luzzatto L, Risitano AM, Notaro R. Paroxysmal nocturnal hemoglobinuria
and eculizumab.Haematologica 2010; 95(4):523-6.
3. Guía clínica HPN. Consenso Español para Diagnóstico y tratamiento de la Hemoglobinuria Paroxística Nocturna. Sociedad Española de HEmatolog{ia y Hemoterapia.SEHH. 2010. Disponible en: http://www.sehh.es/documentos/42/HPN_guia_clinica_v17.pdf
4. Brodsky RA. New inside into Paroxysmal Nocturnal Hemoglobinuria. Hematology (internet). Enero 2006 (citado septiembre 2012); 24(8): [aprox. 5 p]. Disponible en: http://asheducationbook.hematologylibrary.org/content/2006/1/24.full.pdf+htm
5. García-Conde J. Hematología. Madrid: Ediciones Arán; 2003.p.737-42.
6. Parker CJ. Paroxismal Nocturnal Hemoglobinuria. Curr Opin Hematology. 2012;19:141 8. DOI: 10.1097/MOH.0b013c32835/c348.
7. Rodak BF. Hematología. Fundamentos y aplicaciones clínicas.2a ed. Buenos Aires: Editorial Médica Panamericana. 2004; p.286-94
8. Milanés MT, Fernández N, Fundora T, Facundo JC, Hernández P. Hemoglobinuria paroxística nocturna. Actualización. Revista Cubana de Hematología, Inmunología y Hemoterapia (internet). Enero Abril 2003 (citado septiembre 2012); 1(19): [aprox. 5 p.]. Disponible en: http://scielo.sld.cu/scielo.php?script=sci_arttext&pid=S0864 -02892003000100001&lng=es&nrm=iso&tlng=es
9. Rosti V. The molecular basis of paroxysmal nocturnal hemoglobinuria. Haematologica.2000; 85: 82-9.
10. Luzzato L, Bessler M. The dual pathogenesis of paroxysmal nocturnal hemoglobinuria. Curr Opin Hematol.1996;3:101-10.
11. Johnson RJ, Hillmen. Paroxysmal nocturnal hemoglobinuria. Mol Pathol 2002; 55:145-52.
12. Rosse WF. Paroxysmal nocturnal hemoglobinuria. En: Hoffman R, Benz EJ, Shatill SJ, Furie B, Cohen HJ, Silberstein E, McGlave P. Hematology: Basic principles and practice. 3 ed. USA: Churchill Livingstone. 2000; p. 331-42.
13. Yoon JH, Cho HI, Park SS, Chang YH, Kim BK. Mutation analysis of the PIG-A gene in Korean patients with paroxysmal nocturnal haemoglobinuria. J. Clin Pathol. 2002;55:410-3.
14. Prusiner SB, Scott MR, De Armond SJ, Cohen FE. Prion protein biology. Cell 1998;93:337-48.
15. Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease and phospholipase-sensitive. J Biol Chem 1991;266:18217-23.
16. Dodelet VC, Cashman NR. Prion expression in human leukocyte differentiation. Blood 1998;91:1556-61.
17. Meletis J, Terpos E. Paroxysmal nocturnal hemoglobinuria: clinical presentation and association with other haematological disorders. Haema 2001;4:79-88.
18. Richard-Lee G. Hemoglobinuria paroxística nocturna. En: Wintrobe Hematología Clínica. 9 ed. Editorial Inter-Médicos S.A.I.C.I. 1994; p.1072-83.
19. Ray JG, Burows RF, Ginsberg JS, Burrows EA. Paroxysmal nocturnal hemoglobinuria and the risk of venous thrombosis: review and non pregnant patient. Haemostasis 2000;30:103-17.
20. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106:3699709.
21. Hernández-Campo PM, Almeida J, López A, Orfao A. Hemoglobinuria paroxística nocturna. Med Clin(Barc).2008;131:61730.
22. De Latour RP, Mary JY, Salanoubat C,Terriou L,Etienne G,Mothy M, et al. Paroxysmal nocturnal hemoglobinuria: Natural history of disease subcate- gories. Blood.2008; 112(4):3099106.
23. Meletis J, Terpos E, Samarkos M. Detection of CD55 and/or CD59 deficient red cell population in patients with aplastic anemia, myelodysplastic syndrome and myeloproliferative dirsoders. Haematología 2001;31:7-16.
24. Parker C, Omine M, Richards S, Nishimura J, Bessler M, Ware R, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106: 3699709.
25. Richards SJ, Rawstron AC, Hillmen P. Application of flow cytometry to the diagnosis of paroxysmal nocturnal hemoglobinuria. Cytometry Part A. 15 de Agosto 2000 (citado septiembre 2012); 4(42): (aprox. 10 p.). Disponible en: http://onlinelibrary.wiley.com/doi/10.1002/1097-0320%2820000815%2942:4%3C223::AID-CYTO2%3E3.0.CO;2-D/full
26. Richards SJ, Barnett D. The role of flow cytometry in the diagnosis of paroxysmal nocturnal hemoglobinuria in the clinical laboratory.Clin Lab Med. 2007;27: 57790.
27. Risitano Antonio M, Rotoli Bruno. Paroxismal nocturnal hemoglobinuria: pathophysiology, natural history and treatment options in the era of biological agents. Biologics. Junio 2008; 2(2): 205 - 222.
28. Rother RP, Bell L, Hillmen P,Gladwin M T. The clinical sequelae of intravascular hemolysis and extracelular plasma hemoglobin: A novel mechanism of human disease. The Journal of the American Medical Association (JAMA). Abril 2005; 293 (15):1653 - 1662.
29. Brodsky RA. How I treat paroxysmal nocturnal hemoglobinuria. Blood. Abril 2009; 113(26): 6522-6527.
30. Luzzato L, Gianfaldoni G, Notaro R. Management of Paroxysmal Nocturnal Haemoglobinuria: a personal view. Br J Haematol 2011; 153:709-720. doi: 10.1111/j.1365-2141.2011.08690.x
31. Brodsky RA. Stem cell transplantation for paroxysmal nocturnal Hemoglobinuria. Haematologica 2010; 95(6): 855-6.
32. Matos- Fernández NA, Abou Mourad Y, Caceres W. Current Status of allogenis hematopoietic stem cell transplantation for paroxismal nocturnal hemoglobinuria. Biol Blood Marrow Transplant 2009; 15(6):656-61. doi: 10.1016/j.bbmt.2008.12.507
33. Rother RP, Rollins SA, Mojcik CF, Brodsky RA, Bell L. Discovery and development of the complement inhibitor eculizumab for the treatment of paroxysmal nocturnal Hemoglobinuria. Nat Biotech, 2007;25: 1256-64.
34. Kelly R, Arnold L, Richards S, Hill A, Bomken C, Hanley J et al. the management of pregnancy in paroxysmal nocturnal Hemoglobinuria of long term eculizumab. Br J Haematol 2010; 149(3):446-50.
35. Luzatto L, Gianfaldoni G. Recent advances in biological and clinical aspects of paroxysmal nocturnal Hemoglobinuria. Int J Haematol 2006; 84: 104-12
36. Caro P, García Y. Estudio eritrocinético en la hemoglobinuria paroxística nocturna. Sangre 1979;24: 627-32
37. Fundora T, García Y, Roque MC, Vidal H, Ferrer R, González A. Incorporación de la transferrina al eritrón en estados anémicos y policitémicos. Sangre 1991;36: 187-91
38. Parker C. Eculizumab for paroxysmal nocturnal haemoglobinuria. Lancet. 2009;373: 75967.
39. Hillmen P,Muus P, Duhrsen U, Risitano AM, Schubert J, Luzzatto L, et al. Effect on the complement inhibitor eculizumab on thromboembolism in patients with paroxysmal nocturnal hemoglobinuria.Blood.2007;110:41238.
40. Hillmen P. The role of complement inhibition in PNH. Hematology Am Soc Hematol Educ Program.2008; 1:11623.
41. Hill A, Ridley SH, Esser D, Oldroyd RG, Cullen MJ, Kareclas P,et al. Protection of erythrocytes from human complement-mediated lysis by membrane-targeted recombinant solubleCD59:A new approach to PNH therapy.Blood. 2006;107: 21317.
42. Richards SJ, Whitby L,Cullen MJ, Dickinson AJ, Granger V, Reilly JT, et al. Development and evaluation of a stabilized whole-blood preparation as a process control material for screening of paroxysmal nocturnal hemoglobinuria by flow cytometry.CytometryBClinCytom.2009;79B:4755.
43. Wang SA, Pozdyakova O, Jorgensen JL, Medeiros J, Stachurski D, Anderson M, et al. Detection of paroxysmal nocturnal hemoglobinuria clones in patients with myelodysplastic syndromes and related bone marrow diseases, with emphasis on diagnostic pitfalls and caveats.Haematologica.2009; 94: 2937.
44. Algado JT, Luque R, Núñez R, Sánchez B. Infección meningocócica probable en paciente con hemoglobinuria paroxística nocturna y tratamiento con eculizumab. Enferm Infecc Microbiol Clin. 2012;30:109-10.
45. Provan D, Gribben E.J. Ed. Molecular Hematology. 3th ed. Oxford: Wiley Blackwell. 2010.
46. De Guibert S, de Latour RP, Varoqueaux N, Labussiere H, Bernard R, Jaulmes D et al. paroxysmal nocturnal Hemoglobinuria and pregnancy before the eculizumab era: the French experience. Haematologica 2011;96(9): 1276-1283. doi: 10.3324/haematol.2010.037531.
47. Brodsky RA. Young NS, Antonioli E, Risitano AM, Schrezenmeier H, Shubert J, et al. Multicentrc phase 3 study of the complement inhibitor eculizumab for the treatment of patient with paroxysmal nocturnal Hemoglobinuria. Blood. 2008; 11:1840-7
48. Brodsky RA. Paroxysmal nocturnal hemoglobinuria: Stem cell and clonality. Hematology Am Soc Hematol Educ Program.2008; 1:1115. 11.
49. Sutherland DR, Kuek N, Davidson J, Barth D, Chang H, Eo E, et al. Diagnosing PNH with FLAER and multiparameter flow cytometry. Cytometry B Clin Cytom. 2007; 72B:16777.
50. Oelschlaegel U, Besson I, Arnoulet C, Sainty D, Nowak R, Naumann R, et al. A standarized flow cytometric method for screening paroxysmal nocturnal hemoglobinuria (PNH) measuring CD55 and CD59 expression on erythrocytes and granulocytes.Clin Lab Haem.2001;23:8190.
Recibido: 19 de
octubre de 2012.
Aprobado:
21 de noviembre de 2012.
Dra Ivis Macías Pérez. E Mail: rchematologia@infomed.sld.cu
{kind=link}
{kind=link}
{kind=link}